

Deciphering the complexity of Toll-like receptor signaling
- PMID: 20680392
- PMCID: PMC11115668
- DOI: 10.1007/s00018-010-0464-x
Deciphering the complexity of Toll-like receptor signaling
Abstract
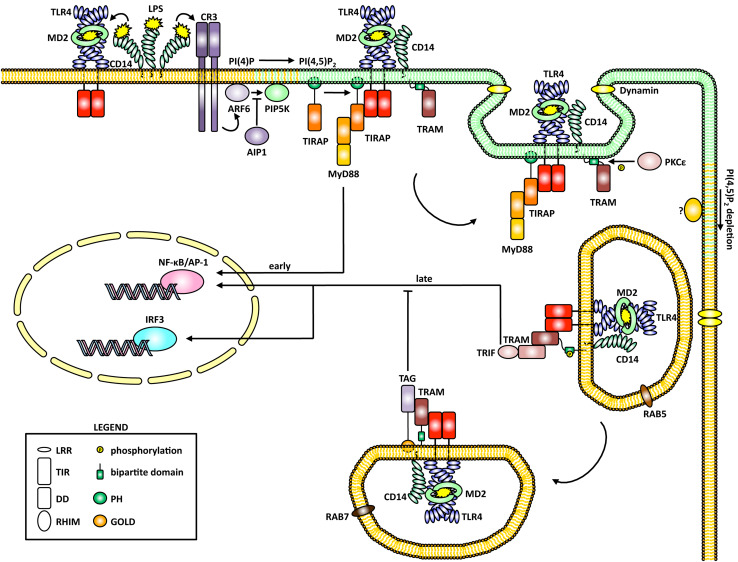
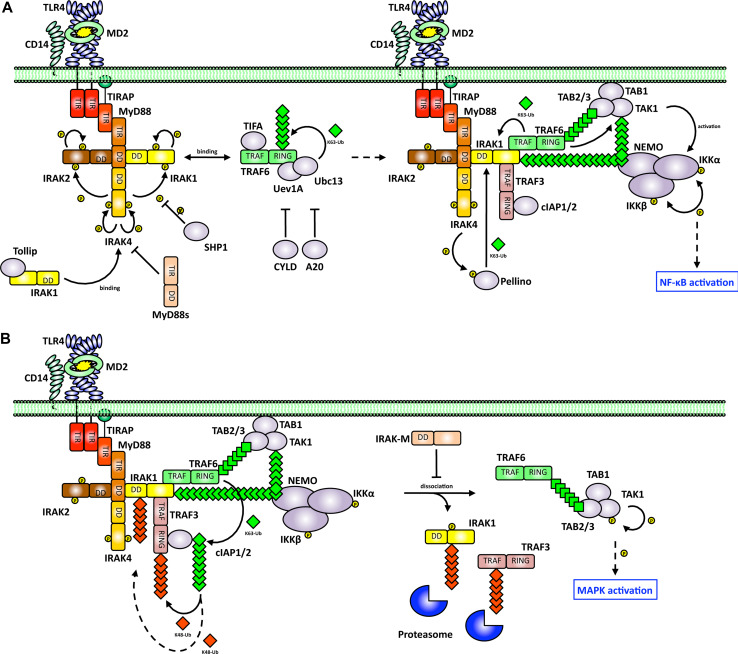
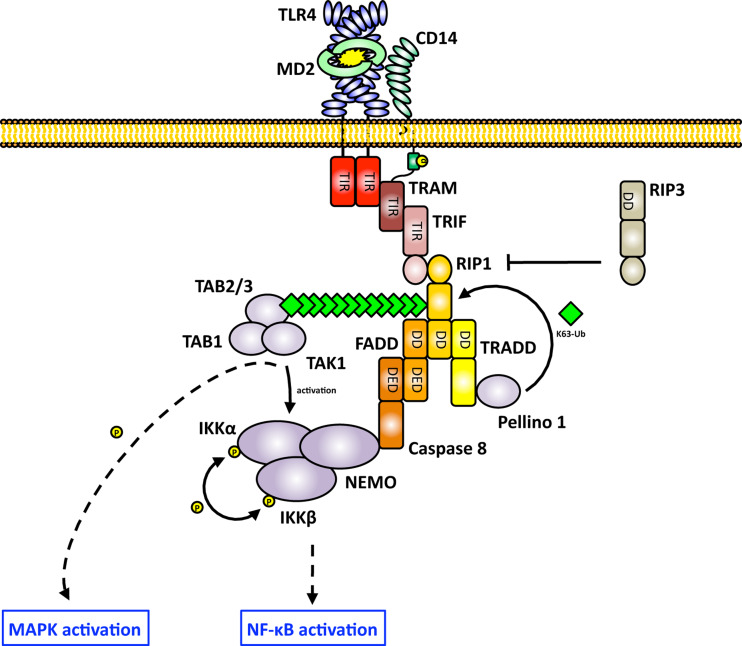
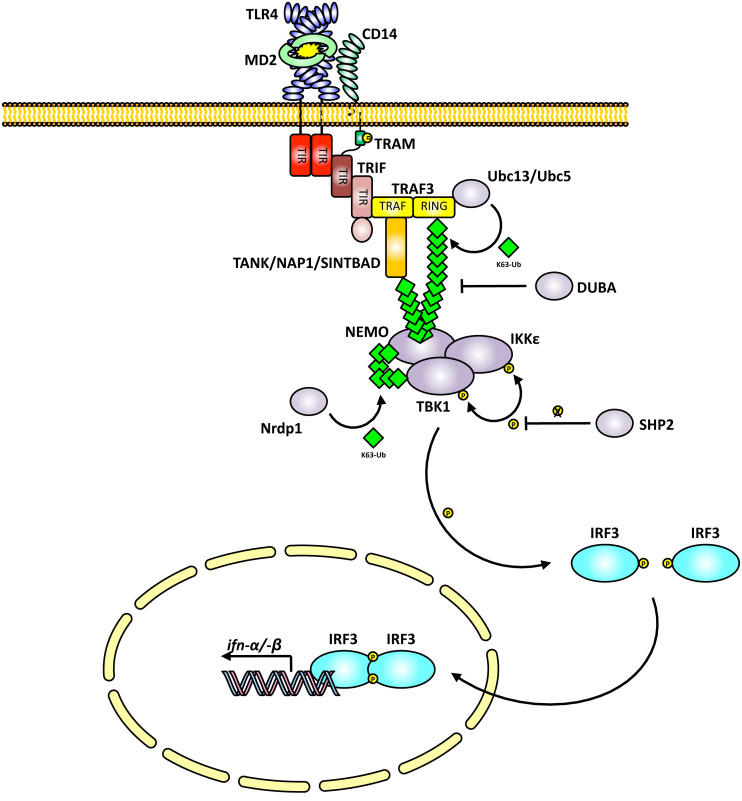
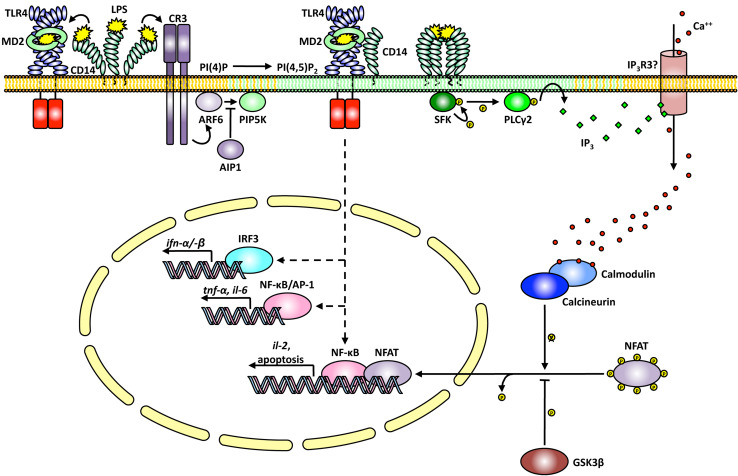
Toll-like receptors (TLRs) are essential players in the innate immune response to invading pathogens. Although extensive research efforts have provided a considerable wealth of information on how TLRs function, substantial gaps in our knowledge still prevent the definition of a complete picture of TLR signaling. However, several recent studies describe additional layers of complexity in the regulation of TLR ligand recognition, adaptor recruitment, posttranslational modifications of signaling proteins, and the newly described, autonomous role of the TLR4 co-receptor CD14. In this review, by using it as model system for the whole TLR family, we attempt to provide a complete description of the signal transduction pathways triggered by TLR4, with a particular emphasis on the molecular and cell biological aspects regulating its function. Finally, we discuss a recently reported model of CD14-dependent signaling and highlight its biological implications.
Figures
Similar articles
-
A role for the adaptor proteins TRAM and TRIF in toll-like receptor 2 signaling.J Biol Chem. 2015 Feb 6;290(6):3209-22. doi: 10.1074/jbc.M114.593426. Epub 2014 Dec 11. J Biol Chem. 2015. PMID: 25505250 Free PMC article.
-
Moraxella catarrhalis activates murine macrophages through multiple toll like receptors and has reduced clearance in lungs from TLR4 mutant mice.PLoS One. 2012;7(5):e37610. doi: 10.1371/journal.pone.0037610. Epub 2012 May 25. PLoS One. 2012. PMID: 22662179 Free PMC article.
-
Endotoxin tolerance dysregulates MyD88- and Toll/IL-1R domain-containing adapter inducing IFN-beta-dependent pathways and increases expression of negative regulators of TLR signaling.J Leukoc Biol. 2009 Oct;86(4):863-75. doi: 10.1189/jlb.0309189. Epub 2009 Aug 5. J Leukoc Biol. 2009. PMID: 19656901 Free PMC article.
-
Toll-like receptors: new players in myocardial ischemia/reperfusion injury.Antioxid Redox Signal. 2011 Oct 1;15(7):1875-93. doi: 10.1089/ars.2010.3723. Epub 2011 Apr 8. Antioxid Redox Signal. 2011. PMID: 21091074 Free PMC article. Review.
-
Role of Toll-like receptor responses for sepsis pathogenesis.Immunobiology. 2007;212(9-10):715-22. doi: 10.1016/j.imbio.2007.09.010. Epub 2007 Nov 26. Immunobiology. 2007. PMID: 18086373 Review.
Cited by
-
Resolvin D2 decreases TLR4 expression to mediate resolution in human monocytes.FASEB J. 2016 Sep;30(9):3181-93. doi: 10.1096/fj.201600375R. Epub 2016 Jun 2. FASEB J. 2016. PMID: 27256622 Free PMC article.
-
Oxidized Low-Density Lipoprotein Induces Apoptosis in Cultured Neonatal Rat Cardiomyocytes by Modulating the TLR4/NF-κB Pathway.Sci Rep. 2016 Jun 9;6:27866. doi: 10.1038/srep27866. Sci Rep. 2016. PMID: 27279424 Free PMC article.
-
Integrin CD11b positively regulates TLR4-induced signalling pathways in dendritic cells but not in macrophages.Nat Commun. 2014;5:3039. doi: 10.1038/ncomms4039. Nat Commun. 2014. PMID: 24423728 Free PMC article.
-
Inositol 1,4,5-trisphosphate 3-kinase B promotes Ca2+ mobilization and the inflammatory activity of dendritic cells.Sci Signal. 2021 Mar 30;14(676):eaaz2120. doi: 10.1126/scisignal.aaz2120. Sci Signal. 2021. PMID: 33785611 Free PMC article.
-
Interaction of antiphospholipid antibodies with endothelial cells in antiphospholipid syndrome.Front Immunol. 2024 Jul 9;15:1361519. doi: 10.3389/fimmu.2024.1361519. eCollection 2024. Front Immunol. 2024. PMID: 39044818 Free PMC article. Review.
References
-
- Janeway CA., Jr Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54:1–13. - PubMed
-
- West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll-like receptors. Annu Rev Cell Dev Biol. 2006;22:409–437. - PubMed
-
- Gay NJ, Gangloff M. Structure and function of Toll receptors and their ligands. Annu Rev Biochem. 2007;76:141–165. - PubMed
-
- Beutler B, Jiang Z, Georgel P, Crozat K, Croker B, Rutschmann S, Du X, Hoebe K. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu Rev Immunol. 2006;24:353–389. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
