

Thalamic gating of corticostriatal signaling by cholinergic interneurons
- PMID: 20670836
- PMCID: PMC4085694
- DOI: 10.1016/j.neuron.2010.06.017
Thalamic gating of corticostriatal signaling by cholinergic interneurons
Abstract
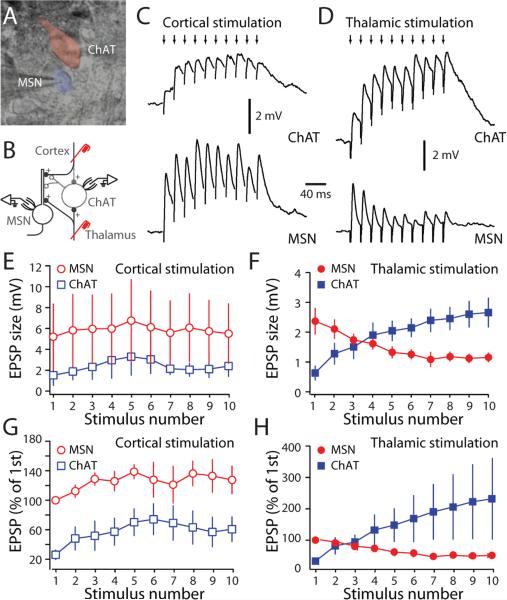
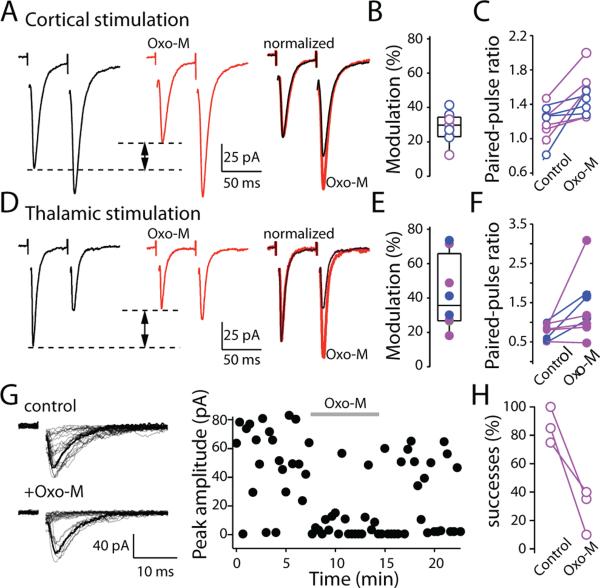
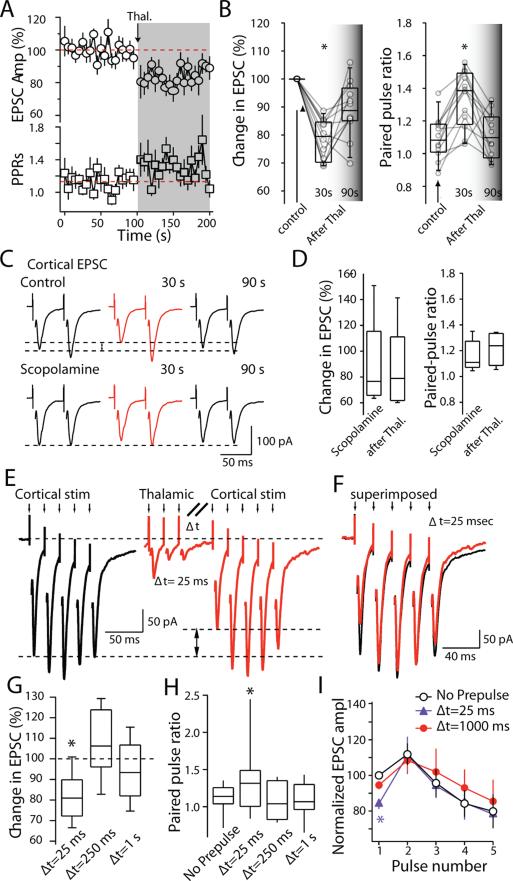
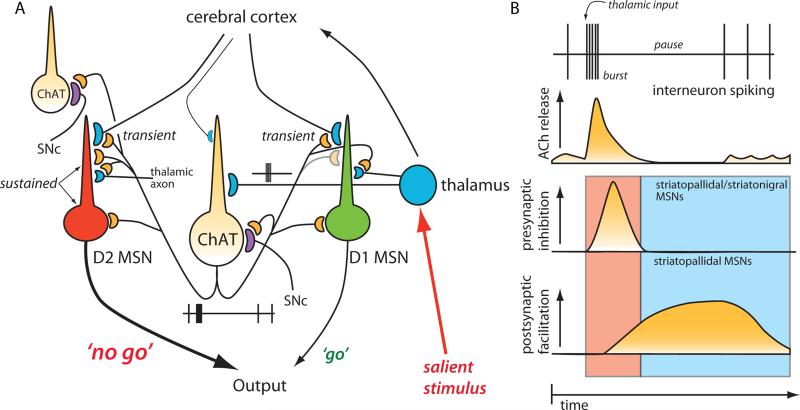
Salient stimuli redirect attention and suppress ongoing motor activity. This attentional shift is thought to rely upon thalamic signals to the striatum to shift cortically driven action selection, but the network mechanisms underlying this interaction are unclear. Using a brain slice preparation that preserved cortico- and thalamostriatal connectivity, it was found that activation of thalamostriatal axons in a way that mimicked the response to salient stimuli induced a burst of spikes in striatal cholinergic interneurons that was followed by a pause lasting more than half a second. This patterned interneuron activity triggered a transient, presynaptic suppression of cortical input to both major classes of principal medium spiny neuron (MSN) that gave way to a prolonged enhancement of postsynaptic responsiveness in striatopallidal MSNs controlling motor suppression. This differential regulation of the corticostriatal circuitry provides a neural substrate for attentional shifts and cessation of ongoing motor activity with the appearance of salient environmental stimuli.
Figures





Comment in
-
Pausing to regroup: thalamic gating of cortico-basal ganglia networks.Neuron. 2010 Jul 29;67(2):175-8. doi: 10.1016/j.neuron.2010.07.010. Neuron. 2010. PMID: 20670826
Similar articles
-
Differential modulation of excitatory and inhibitory striatal synaptic transmission by histamine.J Neurosci. 2011 Oct 26;31(43):15340-51. doi: 10.1523/JNEUROSCI.3144-11.2011. J Neurosci. 2011. PMID: 22031880 Free PMC article.
-
Convergence of cortical and thalamic input to direct and indirect pathway medium spiny neurons in the striatum.Brain Struct Funct. 2014 Sep;219(5):1787-800. doi: 10.1007/s00429-013-0601-z. Epub 2013 Jul 6. Brain Struct Funct. 2014. PMID: 23832596 Free PMC article.
-
Functional comparison of corticostriatal and thalamostriatal postsynaptic responses in striatal neurons of the mouse.Brain Struct Funct. 2018 Apr;223(3):1229-1253. doi: 10.1007/s00429-017-1536-6. Epub 2017 Nov 3. Brain Struct Funct. 2018. PMID: 29101523
-
Excitatory extrinsic afferents to striatal interneurons and interactions with striatal microcircuitry.Eur J Neurosci. 2019 Mar;49(5):593-603. doi: 10.1111/ejn.13881. Epub 2018 Mar 25. Eur J Neurosci. 2019. PMID: 29480942 Free PMC article. Review.
-
Spike-timing dependent plasticity in striatal interneurons.Neuropharmacology. 2011 Apr;60(5):780-8. doi: 10.1016/j.neuropharm.2011.01.023. Epub 2011 Jan 22. Neuropharmacology. 2011. PMID: 21262240 Review.
Cited by
-
GABAergic inputs from direct and indirect striatal projection neurons onto cholinergic interneurons in the primate putamen.J Comp Neurol. 2013 Aug 1;521(11):2502-22. doi: 10.1002/cne.23295. J Comp Neurol. 2013. PMID: 23296794 Free PMC article.
-
Dopamine D2 receptors and striatopallidal transmission in addiction and obesity.Curr Opin Neurobiol. 2013 Aug;23(4):535-8. doi: 10.1016/j.conb.2013.04.012. Epub 2013 May 29. Curr Opin Neurobiol. 2013. PMID: 23726225 Free PMC article. Review.
-
Populations of striatal medium spiny neurons encode vibrotactile frequency in rats: modulation by slow wave oscillations.J Neurophysiol. 2013 Jan;109(2):315-20. doi: 10.1152/jn.00489.2012. Epub 2012 Oct 31. J Neurophysiol. 2013. PMID: 23114217 Free PMC article.
-
The Aggregation of α-Synuclein in the Dorsomedial Striatum Significantly Impairs Cognitive Flexibility in Parkinson's Disease Mice.Biomedicines. 2024 Jul 23;12(8):1634. doi: 10.3390/biomedicines12081634. Biomedicines. 2024. PMID: 39200099 Free PMC article.
-
A spotlight on the elusive striatal cholinergic interneuron.Science. 2021 Apr 23;372(6540):345-346. doi: 10.1126/science.abi4907. Science. 2021. PMID: 33888628 Free PMC article. No abstract available.
References
-
- Akins PT, Surmeier DJ, Kitai ST. M1 muscarinic acetylcholine receptor in cultured rat neostriatum regulates phosphoinositide hydrolysis. J Neurochem. 1990;54:266–273. - PubMed
-
- Alcantara AA, Mrzljak L, Jakab RL, Levey AI, Hersch SM, Goldman-Rakic PS. Muscarinic m1 and m2 receptor proteins in local circuit and projection neurons of the primate striatum: anatomical evidence for cholinergic modulation of glutamatergic prefronto-striatal pathways. J Comp Neurol. 2001;434:445–460. - PubMed
-
- Aosaki T, Graybiel AM, Kimura M. Effect of the nigrostriatal dopamine system on acquired neural responses in the striatum of behaving monkeys. Science. 1994;265:412–415. - PubMed
-
- Barral J, Galarraga E, Bargas J. Muscarinic presynaptic inhibition of neostriatal glutamatergic afferents is mediated by Q-type Ca2+ channels. Brain Res Bull. 1999;49:285–289. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
