

Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus
- PMID: 20664172
- PMCID: PMC2912177
- DOI: 10.1172/JCI37539
Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus
Abstract
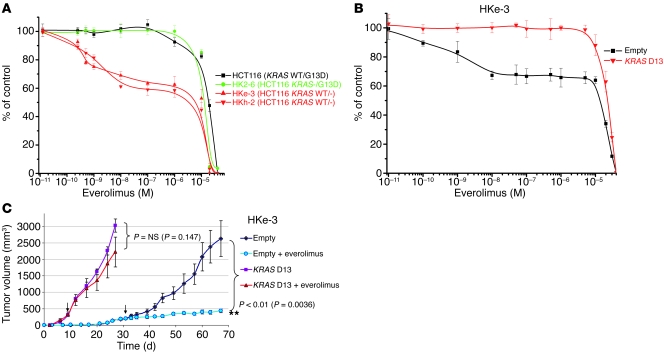
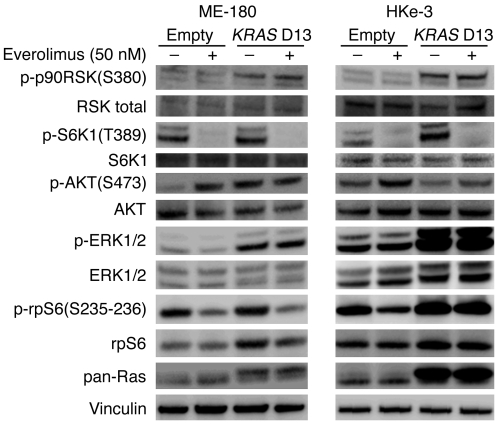
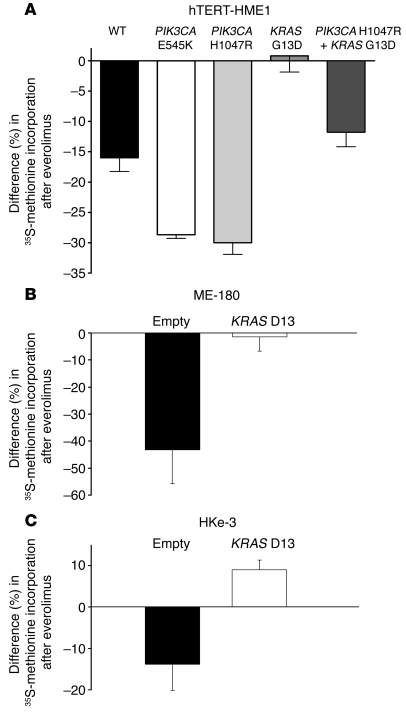
Personalized cancer medicine is based on the concept that targeted therapies are effective on subsets of patients whose tumors carry specific molecular alterations. Several mammalian target of rapamycin (mTOR) inhibitors are in preclinical or clinical trials for cancers, but the molecular basis of sensitivity or resistance to these inhibitors among patients is largely unknown. Here we have identified oncogenic variants of phosphoinositide-3-kinase, catalytic, alpha polypeptide (PIK3CA) and KRAS as determinants of response to the mTOR inhibitor everolimus. Human cancer cells carrying alterations in the PI3K pathway were responsive to everolimus, both in vitro and in vivo, except when KRAS mutations occurred concomitantly or were exogenously introduced. In human cancer cells with mutations in both PIK3CA and KRAS, genetic ablation of mutant KRAS reinstated response to the drug. Consistent with these data, PIK3CA mutant cells, but not KRAS mutant cells, displayed everolimus-sensitive translation. Importantly, in a cohort of metastatic cancer patients, the presence of oncogenic KRAS mutations was associated with lack of benefit after everolimus therapy. Thus, our results demonstrate that alterations in the KRAS and PIK3CA genes may represent biomarkers to optimize treatment of patients with mTOR inhibitors.
Figures


Comment in
-
PIK3CA and KRAS mutations predict for response to everolimus therapy: now that's RAD001.J Clin Invest. 2010 Aug;120(8):2655-8. doi: 10.1172/JCI44026. Epub 2010 Jul 26. J Clin Invest. 2010. PMID: 20664174 Free PMC article.
Similar articles
-
PIK3CA and KRAS mutations predict for response to everolimus therapy: now that's RAD001.J Clin Invest. 2010 Aug;120(8):2655-8. doi: 10.1172/JCI44026. Epub 2010 Jul 26. J Clin Invest. 2010. PMID: 20664174 Free PMC article.
-
Translational studies within the TAMRAD randomized GINECO trial: evidence for mTORC1 activation marker as a predictive factor for everolimus efficacy in advanced breast cancer.Ann Oncol. 2015 Jan;26(1):120-125. doi: 10.1093/annonc/mdu497. Epub 2014 Oct 31. Ann Oncol. 2015. PMID: 25361980 Clinical Trial.
-
Dependence on phosphoinositide 3-kinase and RAS-RAF pathways drive the activity of RAF265, a novel RAF/VEGFR2 inhibitor, and RAD001 (Everolimus) in combination.Mol Cancer Ther. 2010 Feb;9(2):358-68. doi: 10.1158/1535-7163.MCT-09-1014. Epub 2010 Feb 2. Mol Cancer Ther. 2010. PMID: 20124452
-
Impact of genetic alterations on mTOR-targeted cancer therapy.Chin J Cancer. 2013 May;32(5):270-4. doi: 10.5732/cjc.013.10005. Epub 2013 Mar 15. Chin J Cancer. 2013. PMID: 23489586 Free PMC article. Review.
-
New inhibitors of the mammalian target of rapamycin signaling pathway for cancer.Expert Opin Investig Drugs. 2010 Aug;19(8):919-30. doi: 10.1517/13543784.2010.499121. Expert Opin Investig Drugs. 2010. PMID: 20569080 Review.
Cited by
-
Mutations in critical domains confer the human mTOR gene strong tumorigenicity.J Biol Chem. 2013 Mar 1;288(9):6511-21. doi: 10.1074/jbc.M112.399485. Epub 2013 Jan 15. J Biol Chem. 2013. PMID: 23322780 Free PMC article.
-
Genomic Determinants of PI3K Pathway Inhibitor Response in Cancer.Front Oncol. 2012 Aug 31;2:109. doi: 10.3389/fonc.2012.00109. eCollection 2012. Front Oncol. 2012. PMID: 22970424 Free PMC article.
-
Predicting everolimus treatment efficacy in patients with advanced endometrial carcinoma: a GINECO group study.Target Oncol. 2013 Dec;8(4):243-51. doi: 10.1007/s11523-012-0242-9. Epub 2012 Dec 13. Target Oncol. 2013. PMID: 23238879 Clinical Trial.
-
Specific inhibition of p110α subunit of PI3K: putative therapeutic strategy for KRAS mutant colorectal cancers.Oncotarget. 2016 Oct 18;7(42):68546-68558. doi: 10.18632/oncotarget.11843. Oncotarget. 2016. PMID: 27602501 Free PMC article.
-
Landscape of clinically actionable mutations in breast cancer 'A cohort study'.Transl Oncol. 2021 Jan;14(1):100877. doi: 10.1016/j.tranon.2020.100877. Epub 2020 Oct 21. Transl Oncol. 2021. PMID: 33099186 Free PMC article.
References
-
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988–1004. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
