

Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling
- PMID: 20616044
- PMCID: PMC2906569
- DOI: 10.1073/pnas.1000132107
Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling
Abstract
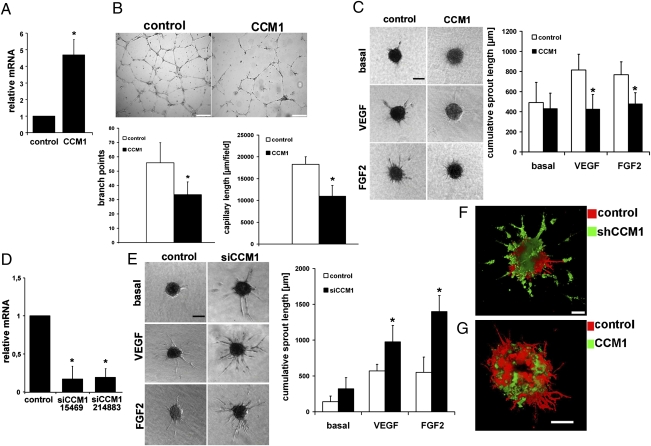
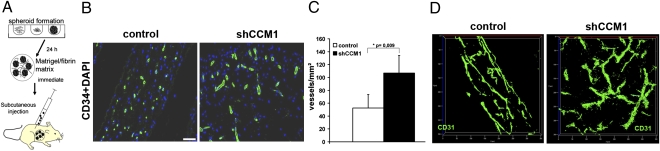
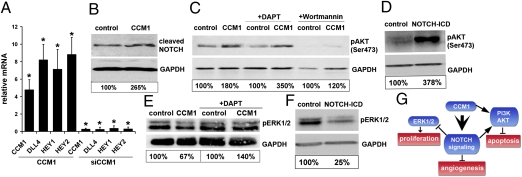
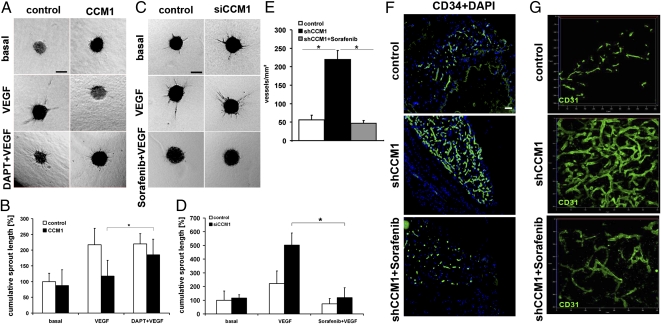
Cerebral cavernous malformations (CCM) are frequent vascular abnormalities caused by mutations in one of the CCM genes. CCM1 (also known as KRIT1) stabilizes endothelial junctions and is essential for vascular morphogenesis in mouse embryos. However, cellular functions of CCM1 during the early steps of the CCM pathogenesis remain unknown. We show here that CCM1 represents an antiangiogenic protein to keep the human endothelium quiescent. CCM1 inhibits endothelial proliferation, apoptosis, migration, lumen formation, and sprouting angiogenesis in primary human endothelial cells. CCM1 strongly induces DLL4-NOTCH signaling, which promotes AKT phosphorylation but reduces phosphorylation of the mitogen-activated protein kinase ERK. Consistently, blocking of NOTCH activity alleviates CCM1 effects. ERK phosphorylation is increased in human CCM lesions. Transplantation of CCM1-silenced human endothelial cells into SCID mice recapitulates hallmarks of the CCM pathology and serves as a unique CCM model system. In this setting, the multikinase inhibitor Sorafenib can ameliorate loss of CCM1-induced excessive microvascular growth, reducing the microvessel density to levels of normal wild-type endothelial cells. Collectively, our data suggest that the origin of CCM lesions is caused by perturbed Notch signaling-induced excessive capillary sprouting, which can be therapeutically targeted.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Concepts and hypothesis: integrin cytoplasmic domain-associated protein-1 (ICAP-1) as a potential player in cerebral cavernous malformation.J Neurol. 2013 Jan;260(1):10-9. doi: 10.1007/s00415-012-6567-6. Epub 2012 Jun 19. J Neurol. 2013. PMID: 22711159 Review.
-
Integrin cytoplasmic domain-associated protein-1 attenuates sprouting angiogenesis.Circ Res. 2010 Sep 3;107(5):592-601. doi: 10.1161/CIRCRESAHA.110.217257. Epub 2010 Jul 8. Circ Res. 2010. PMID: 20616313
-
Loss of CCM3 impairs DLL4-Notch signalling: implication in endothelial angiogenesis and in inherited cerebral cavernous malformations.J Cell Mol Med. 2013 Mar;17(3):407-18. doi: 10.1111/jcmm.12022. Epub 2013 Feb 7. J Cell Mol Med. 2013. PMID: 23388056 Free PMC article.
-
A novel mouse model of cerebral cavernous malformations based on the two-hit mutation hypothesis recapitulates the human disease.Hum Mol Genet. 2011 Jan 15;20(2):211-22. doi: 10.1093/hmg/ddq433. Epub 2010 Oct 11. Hum Mol Genet. 2011. PMID: 20940147 Free PMC article.
-
Role of Delta-Notch signaling in cerebral cavernous malformations.Neurosurg Rev. 2016 Oct;39(4):581-9. doi: 10.1007/s10143-015-0699-y. Epub 2016 Jan 16. Neurosurg Rev. 2016. PMID: 26779617 Review.
Cited by
-
Concepts and hypothesis: integrin cytoplasmic domain-associated protein-1 (ICAP-1) as a potential player in cerebral cavernous malformation.J Neurol. 2013 Jan;260(1):10-9. doi: 10.1007/s00415-012-6567-6. Epub 2012 Jun 19. J Neurol. 2013. PMID: 22711159 Review.
-
SYNJ2BP inhibits tumor growth and metastasis by activating DLL4 pathway in hepatocellular carcinoma.J Exp Clin Cancer Res. 2016 Jul 20;35(1):115. doi: 10.1186/s13046-016-0385-0. J Exp Clin Cancer Res. 2016. PMID: 27440153 Free PMC article.
-
Venous malformation vessels are improperly specified and hyperproliferative.PLoS One. 2021 May 27;16(5):e0252342. doi: 10.1371/journal.pone.0252342. eCollection 2021. PLoS One. 2021. PMID: 34043714 Free PMC article.
-
Beyond multiple mechanisms and a unique drug: Defective autophagy as pivotal player in cerebral cavernous malformation pathogenesis and implications for targeted therapies.Rare Dis. 2016 Jan 25;4(1):e1142640. doi: 10.1080/21675511.2016.1142640. eCollection 2016. Rare Dis. 2016. PMID: 27141412 Free PMC article.
-
Notch signaling in cerebrovascular diseases (Review).Mol Med Rep. 2016 Oct;14(4):2883-98. doi: 10.3892/mmr.2016.5641. Epub 2016 Aug 19. Mol Med Rep. 2016. PMID: 27574001 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
