

Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment
- PMID: 20570886
- PMCID: PMC3436125
- DOI: 10.1158/0008-5472.CAN-09-2356
Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment
Abstract
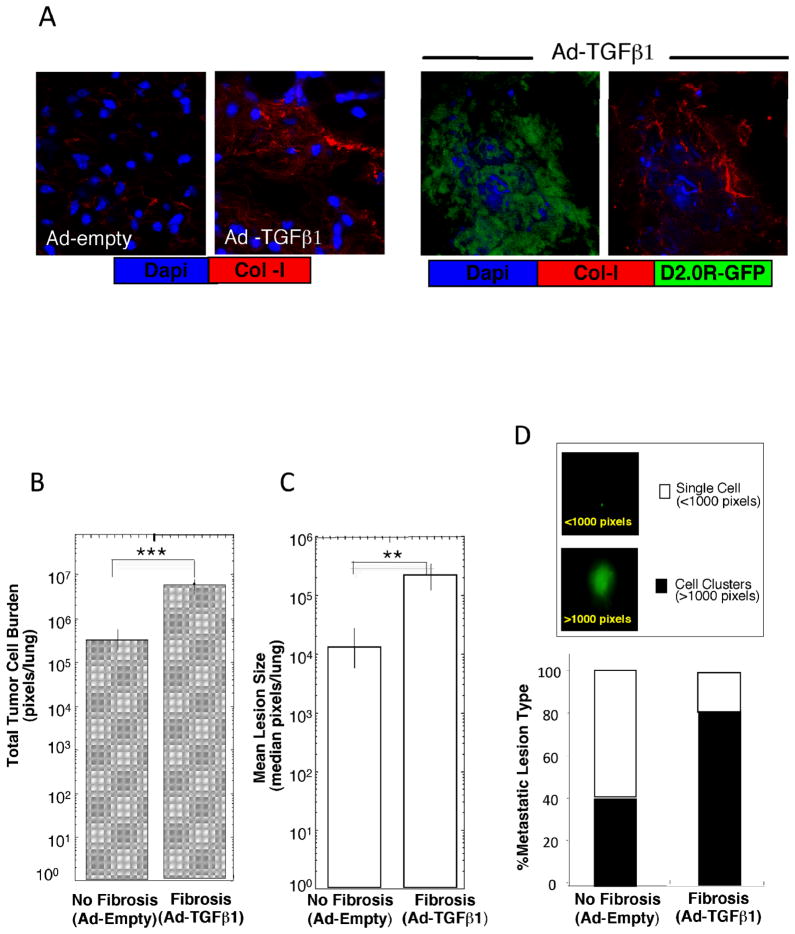
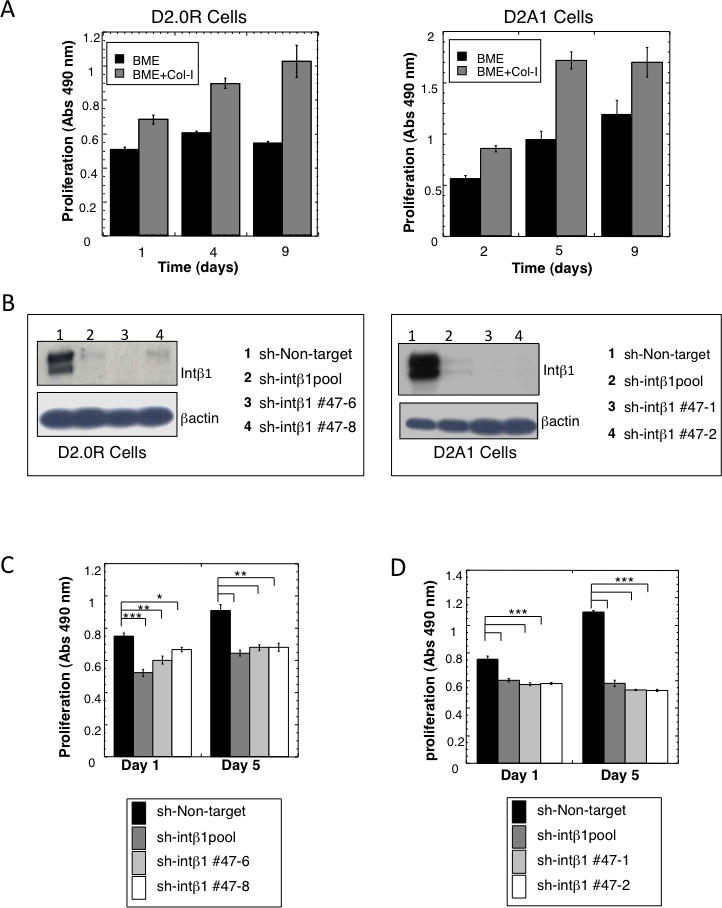
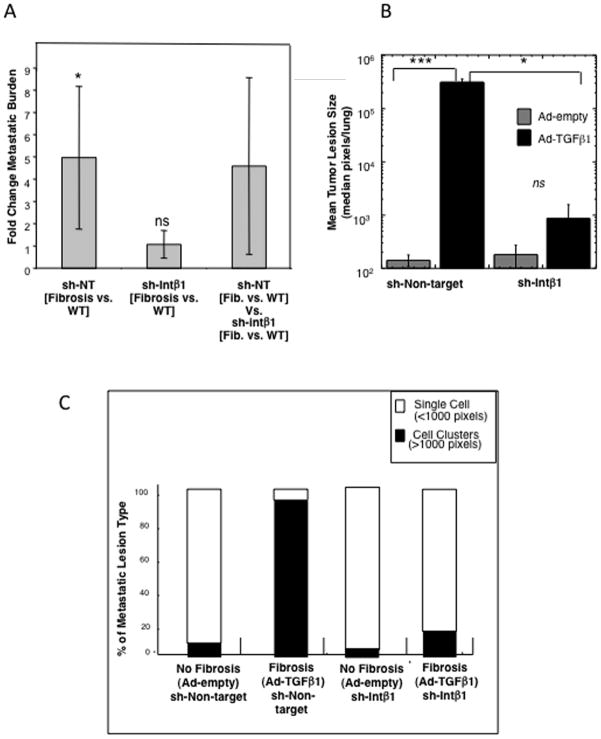
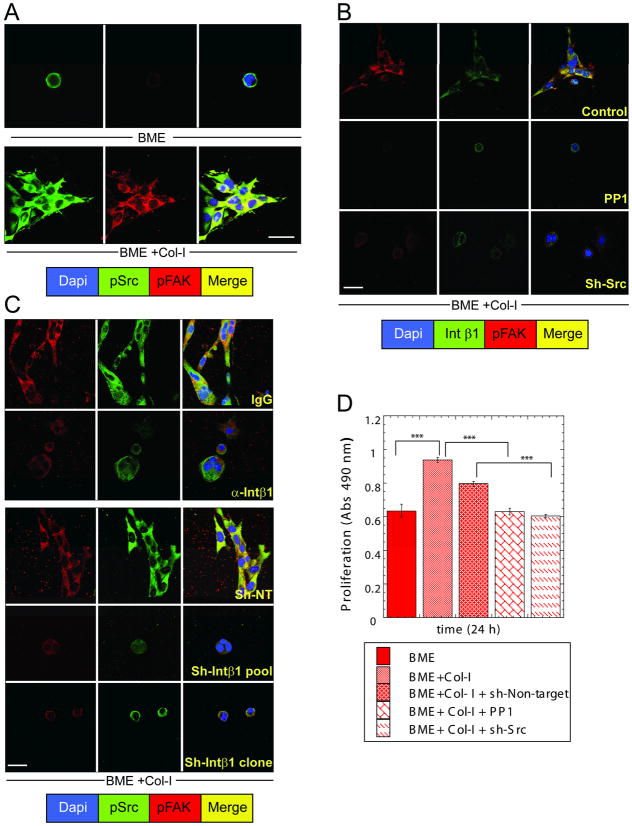
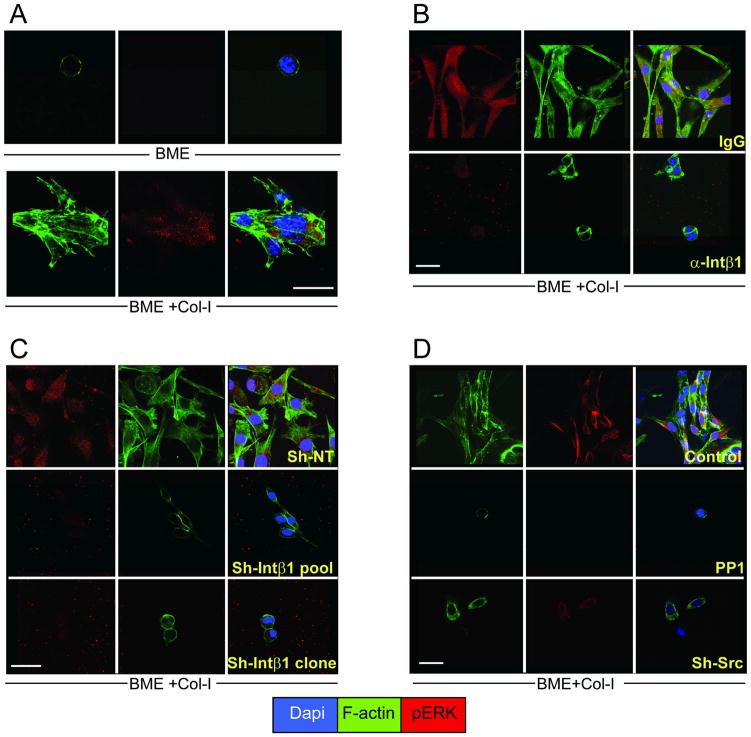
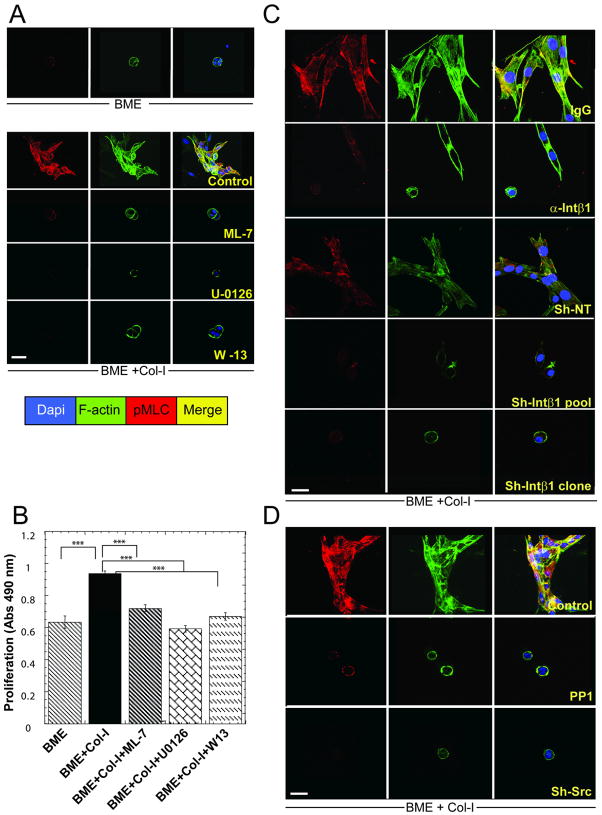
Breast cancer that recurs as metastatic disease many years after primary tumor resection and adjuvant therapy seems to arise from tumor cells that disseminated early in the course of disease but did not develop into clinically apparent lesions. These long-term surviving, disseminated tumor cells maintain a state of dormancy, but may be triggered to proliferate through largely unknown factors. We now show that the induction of fibrosis, associated with deposition of type I collagen (Col-I) in the in vivo metastatic microenvironment, induces dormant D2.0R cells to form proliferative metastatic lesions through beta1-integrin signaling. In vitro studies using a three-dimensional culture system modeling dormancy showed that Col-I induces quiescent D2.0R cells to proliferate through beta1-integrin activation of SRC and focal adhesion kinase, leading to extracellular signal-regulated kinase (ERK)-dependent myosin light chain phosphorylation by myosin light chain kinase and actin stress fiber formation. Blocking beta1-integrin, Src, ERK, or myosin light chain kinase by short hairpin RNA or pharmacologic approaches inhibited Col-I-induced activation of this signaling cascade, cytoskeletal reorganization, and proliferation. These findings show that fibrosis with Col-I enrichment at the metastatic site may be a critical determinant of cytoskeletal reorganization in dormant tumor cells, leading to their transition from dormancy to metastatic growth. Thus, inhibiting Col-I production, its interaction with beta1-integrin, and downstream signaling of beta1-integrin may be important strategies for preventing or treating recurrent metastatic disease.
(c)2010 AACR.
Conflict of interest statement
The authors have no conflicts to disclose.
Figures
Similar articles
-
Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton.Cancer Res. 2008 Aug 1;68(15):6241-50. doi: 10.1158/0008-5472.CAN-07-6849. Cancer Res. 2008. PMID: 18676848 Free PMC article.
-
TGF-β stimulates Pyk2 expression as part of an epithelial-mesenchymal transition program required for metastatic outgrowth of breast cancer.Oncogene. 2013 Apr 18;32(16):2005-15. doi: 10.1038/onc.2012.230. Epub 2012 Jun 18. Oncogene. 2013. PMID: 22710711 Free PMC article.
-
Simvastatin ameliorates altered mechanotransduction in uterine leiomyoma cells.Am J Obstet Gynecol. 2020 Nov;223(5):733.e1-733.e14. doi: 10.1016/j.ajog.2020.05.012. Epub 2020 May 15. Am J Obstet Gynecol. 2020. PMID: 32417359 Free PMC article.
-
Regulation of dormancy during tumor dissemination: the role of the ECM.Cancer Metastasis Rev. 2023 Mar;42(1):99-112. doi: 10.1007/s10555-023-10094-2. Epub 2023 Feb 21. Cancer Metastasis Rev. 2023. PMID: 36802311 Free PMC article. Review.
-
Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth.Eur J Cancer. 2010 May;46(7):1181-8. doi: 10.1016/j.ejca.2010.02.027. Epub 2010 Mar 19. Eur J Cancer. 2010. PMID: 20304630 Free PMC article. Review.
Cited by
-
Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy.Nature. 2021 Jun;594(7864):566-571. doi: 10.1038/s41586-021-03614-z. Epub 2021 Jun 2. Nature. 2021. PMID: 34079127
-
Breast Cancer Metastatic Dormancy and Relapse: An Enigma of Microenvironment(s).Cancer Res. 2022 Dec 16;82(24):4497-4510. doi: 10.1158/0008-5472.CAN-22-1902. Cancer Res. 2022. PMID: 36214624 Free PMC article. Review.
-
Thorny ground, rocky soil: Tissue-specific mechanisms of tumor dormancy and relapse.Semin Cancer Biol. 2022 Jan;78:104-123. doi: 10.1016/j.semcancer.2021.05.007. Epub 2021 May 9. Semin Cancer Biol. 2022. PMID: 33979673 Free PMC article. Review.
-
Matrix rigidity regulates cancer cell growth by modulating cellular metabolism and protein synthesis.PLoS One. 2012;7(5):e37231. doi: 10.1371/journal.pone.0037231. Epub 2012 May 18. PLoS One. 2012. PMID: 22623999 Free PMC article.
-
FGFR1 Signaling Facilitates Obesity-Driven Pulmonary Outgrowth in Metastatic Breast Cancer.Mol Cancer Res. 2024 Mar 1;22(3):254-267. doi: 10.1158/1541-7786.MCR-23-0955. Mol Cancer Res. 2024. PMID: 38153436 Free PMC article.
References
-
- Pantel K, Schlimok G, Braun S, et al. Differential expression of proliferation-associated molecules in individual micrometastatic carcinoma cells. J Natl Cancer Inst. 1993;85:1419–24. - PubMed
-
- Braun S, Vogl FD, Naume B, et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N Engl J Med. 2005;353:793–802. - PubMed
-
- Townson JL, Chambers AF. Dormancy of solitary metastatic cells. Cell Cycle. 2006;5:1744–50. - PubMed
-
- Wikman H, Vessella R, Pantel K. Cancer micrometastasis and tumour dormancy. APMIS. 2008;116:754–70. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
