

Angiotensin-converting enzyme inhibition down-regulates the pro-atherogenic chemokine receptor 9 (CCR9)-chemokine ligand 25 (CCL25) axis
- PMID: 20504763
- PMCID: PMC2906340
- DOI: 10.1074/jbc.M110.117481
Angiotensin-converting enzyme inhibition down-regulates the pro-atherogenic chemokine receptor 9 (CCR9)-chemokine ligand 25 (CCL25) axis
Abstract
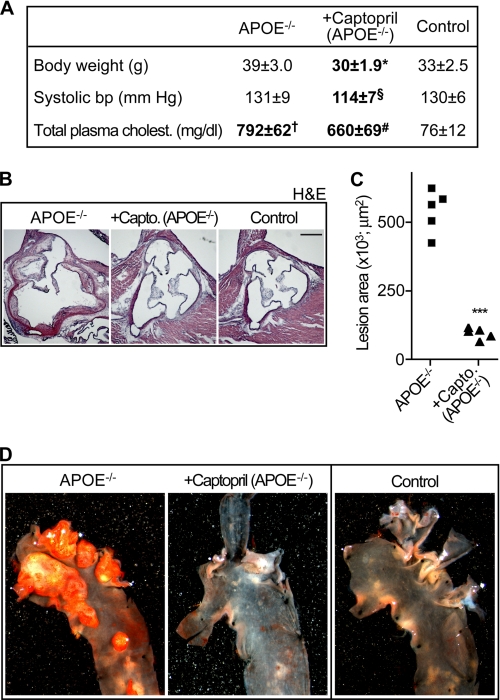
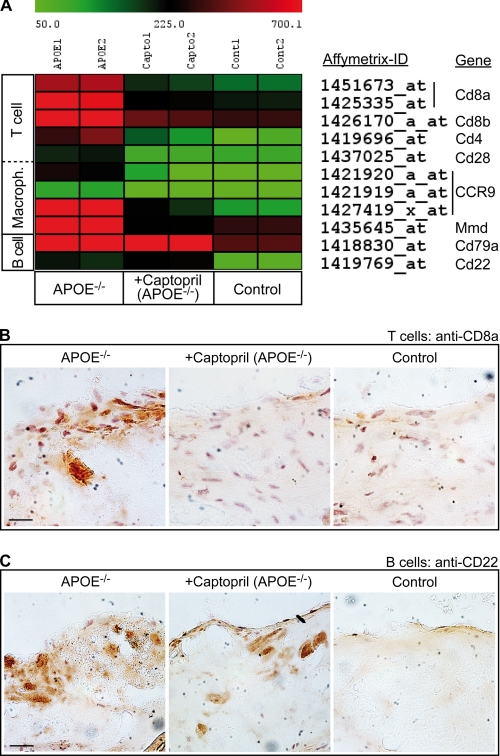
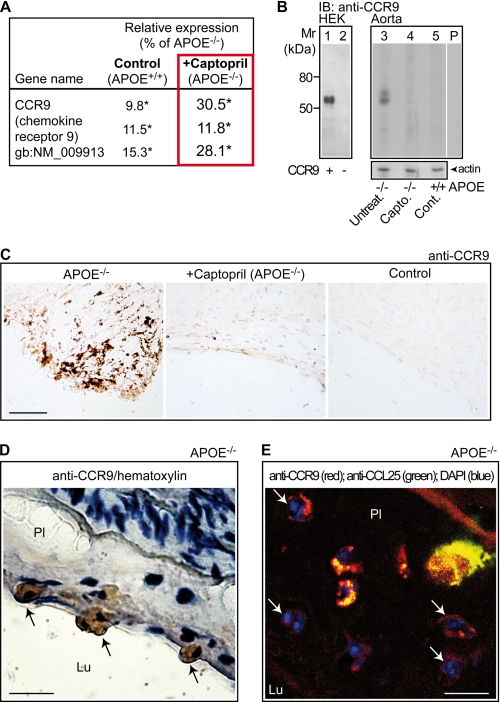
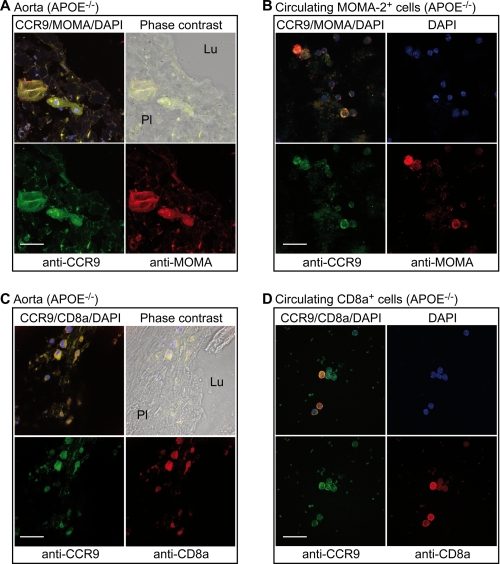
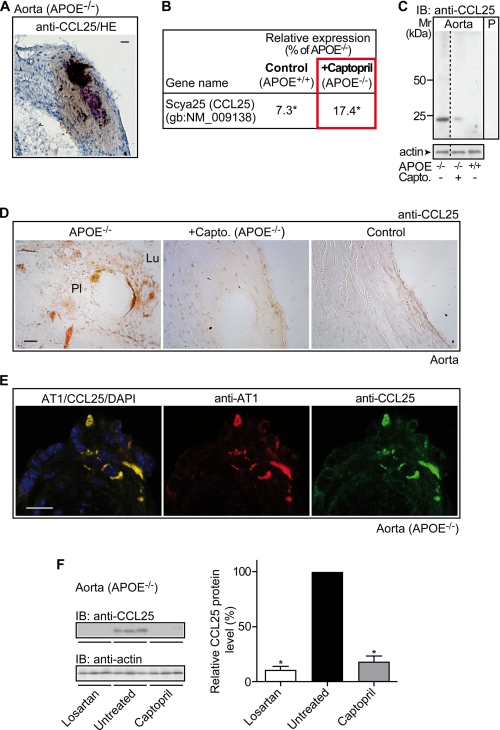
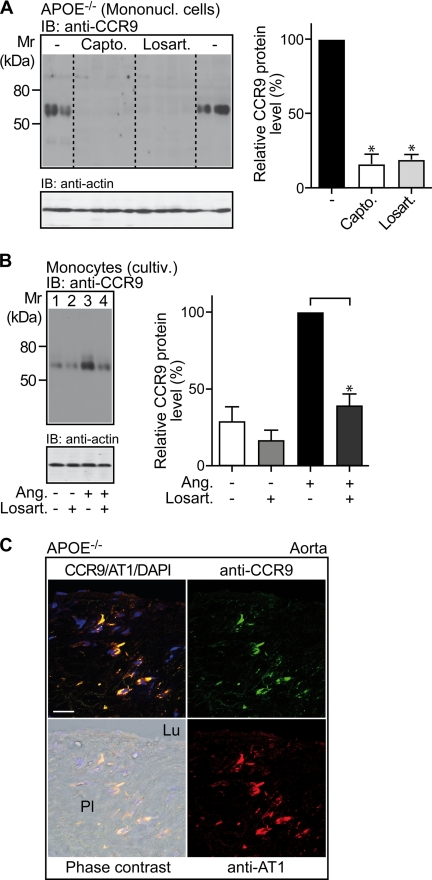
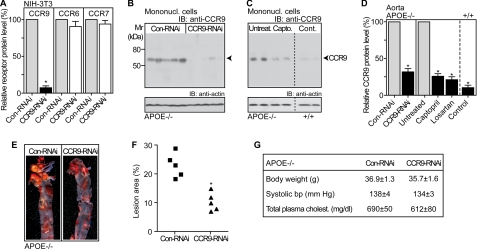
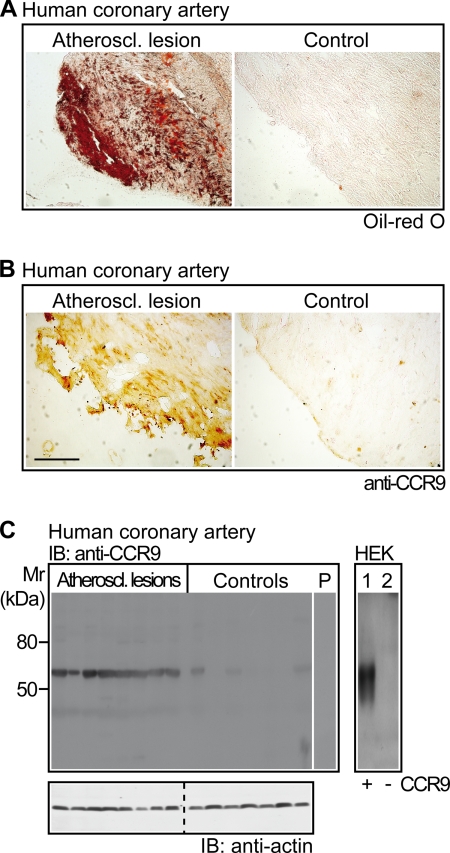
Many experimental and clinical studies suggest a relationship between enhanced angiotensin II release by the angiotensin-converting enzyme (ACE) and the pathophysiology of atherosclerosis. The atherosclerosis-enhancing effects of angiotensin II are complex and incompletely understood. To identify anti-atherogenic target genes, we performed microarray gene expression profiling of the aorta during atherosclerosis prevention with the ACE inhibitor, captopril. Atherosclerosis-prone apolipoprotein E (apoE)-deficient mice were used as a model to decipher susceptible genes regulated during atherosclerosis prevention with captopril. Microarray gene expression profiling and immunohistology revealed that captopril treatment for 7 months strongly decreased the recruitment of pro-atherogenic immune cells into the aorta. Captopril-mediated inhibition of plaque-infiltrating immune cells involved down-regulation of the C-C chemokine receptor 9 (CCR9). Reduced cell migration correlated with decreased numbers of aorta-resident cells expressing the CCR9-specific chemoattractant factor, chemokine ligand 25 (CCL25). The CCL25-CCR9 axis was pro-atherogenic, because inhibition of CCR9 by RNA interference in hematopoietic progenitors of apoE-deficient mice significantly retarded the development of atherosclerosis. Analysis of coronary artery biopsy specimens of patients with coronary artery atherosclerosis undergoing bypass surgery also showed strong infiltrates of CCR9-positive cells in atherosclerotic lesions. Thus, the C-C chemokine receptor, CCR9, exerts a significant role in atherosclerosis.
Figures
Similar articles
-
Deficiency in lymphotoxin β receptor protects from atherosclerosis in apoE-deficient mice.Circ Res. 2015 Apr 10;116(8):e57-68. doi: 10.1161/CIRCRESAHA.116.305723. Epub 2015 Mar 4. Circ Res. 2015. PMID: 25740843
-
Role of beta7 integrin and the chemokine/chemokine receptor pair CCL25/CCR9 in modeled TNF-dependent Crohn's disease.Gastroenterology. 2008 Jun;134(7):2025-35. doi: 10.1053/j.gastro.2008.02.085. Epub 2008 Mar 5. Gastroenterology. 2008. PMID: 18439426
-
Monocytes/macrophages express chemokine receptor CCR9 in rheumatoid arthritis and CCL25 stimulates their differentiation.Arthritis Res Ther. 2010;12(4):R161. doi: 10.1186/ar3120. Epub 2010 Aug 25. Arthritis Res Ther. 2010. PMID: 20738854 Free PMC article.
-
CCR9 and CCL25: A review of their roles in tumor promotion.J Cell Physiol. 2020 Dec;235(12):9121-9132. doi: 10.1002/jcp.29782. Epub 2020 May 13. J Cell Physiol. 2020. PMID: 32401349 Review.
-
The Roles of CCR9/CCL25 in Inflammation and Inflammation-Associated Diseases.Front Cell Dev Biol. 2021 Aug 19;9:686548. doi: 10.3389/fcell.2021.686548. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34490243 Free PMC article. Review.
Cited by
-
C-C Motif Chemokine Receptor 9 Exacerbates Pressure Overload-Induced Cardiac Hypertrophy and Dysfunction.J Am Heart Assoc. 2016 May 4;5(5):e003342. doi: 10.1161/JAHA.116.003342. J Am Heart Assoc. 2016. PMID: 27146447 Free PMC article.
-
Immunomodulatory Activity of the Most Commonly Used Antihypertensive Drugs-Angiotensin Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers.Int J Mol Sci. 2022 Feb 4;23(3):1772. doi: 10.3390/ijms23031772. Int J Mol Sci. 2022. PMID: 35163696 Free PMC article. Review.
-
Inhibition of G-protein-coupled receptor kinase 2 (GRK2) triggers the growth-promoting mitogen-activated protein kinase (MAPK) pathway.J Biol Chem. 2013 Mar 15;288(11):7738-7755. doi: 10.1074/jbc.M112.428078. Epub 2013 Jan 28. J Biol Chem. 2013. PMID: 23362259 Free PMC article.
-
ILC2-mediated immune crosstalk in chronic (vascular) inflammation.Front Immunol. 2023 Dec 20;14:1326440. doi: 10.3389/fimmu.2023.1326440. eCollection 2023. Front Immunol. 2023. PMID: 38179045 Free PMC article. Review.
-
Discovery of crucial cytokines associated with abdominal aortic aneurysm formation by protein array analysis.Exp Biol Med (Maywood). 2019 Dec;244(18):1648-1657. doi: 10.1177/1535370219885101. Epub 2019 Oct 31. Exp Biol Med (Maywood). 2019. PMID: 31665916 Free PMC article.
References
-
- Lonn E., Yusuf S., Dzavik V., Doris C., Yi Q., Smith S., Moore-Cox A., Bosch J., Riley W., Teo K.SECURE Investigators (2001) Circulation 103, 919–925 - PubMed
-
- Olsen M. H., Wachtell K., Neland K., Bella J. N., Rokkedal J., Dige-Petersen H., Ibsen H. (2005) Blood Press 14, 177–183 - PubMed
-
- Yusuf S., Sleight P., Pogue J., Bosch J., Davies R., Dagenais G. (2000) New. Engl. J. Med. 342, 145–153 - PubMed
-
- Stumpe K. O., Agabiti-Rosei E., Zielinski T., Schremmer D., Scholze J., Laeis P., Schwandt P., Ludwig M.MORE study investigators (2007) Ther. Adv. Cardiovasc. Dis. 1, 97–106 - PubMed
-
- Wassmann S., Czech T., van Eickels M., Fleming I., Böhm M., Nickenig G. (2004) Circulation 110, 3062–3067 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
