

Tau Ser262 phosphorylation is critical for Abeta42-induced tau toxicity in a transgenic Drosophila model of Alzheimer's disease
- PMID: 20466736
- PMCID: PMC2901137
- DOI: 10.1093/hmg/ddq200
Tau Ser262 phosphorylation is critical for Abeta42-induced tau toxicity in a transgenic Drosophila model of Alzheimer's disease
Abstract
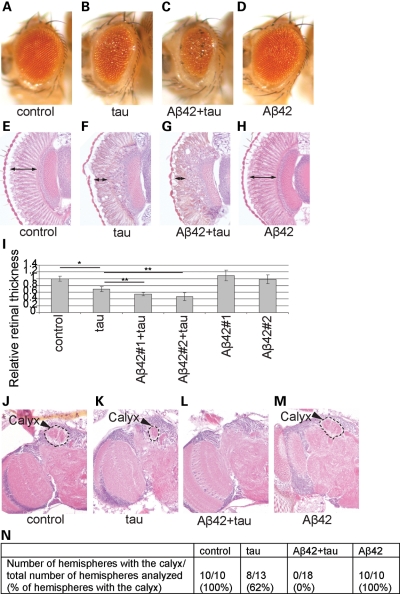
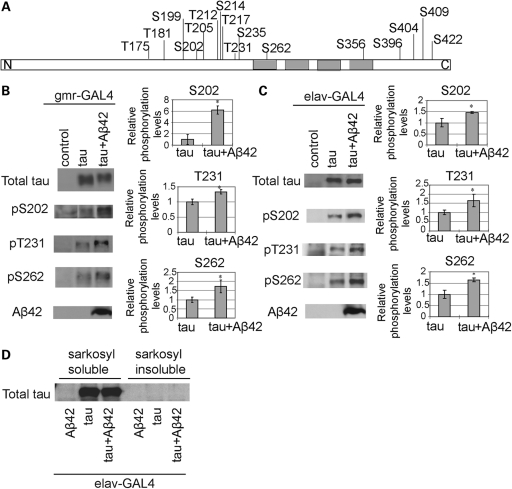
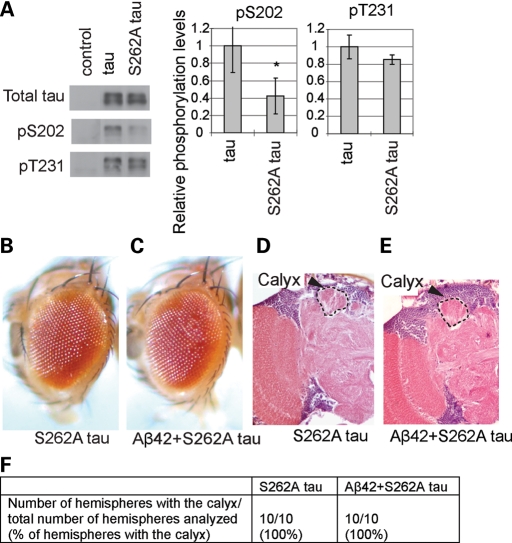
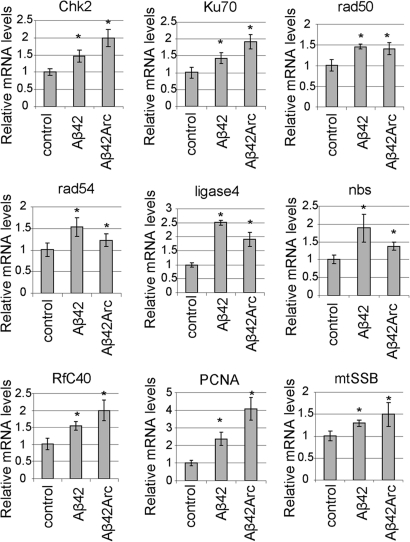
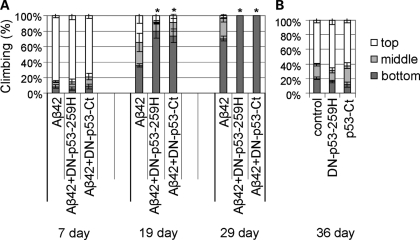
The amyloid-beta 42 (Abeta42) peptide has been suggested to promote tau phosphorylation and toxicity in Alzheimer's disease (AD) pathogenesis; however, the underlying mechanisms are not fully understood. Using transgenic Drosophila expressing both human Abeta42 and tau, we show here that tau phosphorylation at Ser262 plays a critical role in Abeta42-induced tau toxicity. Co-expression of Abeta42 increased tau phosphorylation at AD-related sites including Ser262, and enhanced tau-induced neurodegeneration. In contrast, formation of either sarkosyl-insoluble tau or paired helical filaments was not induced by Abeta42. Co-expression of Abeta42 and tau carrying the non-phosphorylatable Ser262Ala mutation did not cause neurodegeneration, suggesting that the Ser262 phosphorylation site is required for the pathogenic interaction between Abeta42 and tau. We have recently reported that the DNA damage-activated Checkpoint kinase 2 (Chk2) phosphorylates tau at Ser262 and enhances tau toxicity in a transgenic Drosophila model. We detected that expression of Chk2, as well as a number of genes involved in DNA repair pathways, was increased in the Abeta42 fly brains. The induction of a DNA repair response is protective against Abeta42 toxicity, since blocking the function of the tumor suppressor p53, a key transcription factor for the induction of DNA repair genes, in neurons exacerbated Abeta42-induced neuronal dysfunction. Our results demonstrate that tau phosphorylation at Ser262 is crucial for Abeta42-induced tau toxicity in vivo, and suggest a new model of AD progression in which activation of DNA repair pathways is protective against Abeta42 toxicity but may trigger tau phosphorylation and toxicity in AD pathogenesis.
Figures
Similar articles
-
A DNA damage-activated checkpoint kinase phosphorylates tau and enhances tau-induced neurodegeneration.Hum Mol Genet. 2010 May 15;19(10):1930-8. doi: 10.1093/hmg/ddq068. Epub 2010 Feb 16. Hum Mol Genet. 2010. PMID: 20159774 Free PMC article.
-
Stabilization of Microtubule-Unbound Tau via Tau Phosphorylation at Ser262/356 by Par-1/MARK Contributes to Augmentation of AD-Related Phosphorylation and Aβ42-Induced Tau Toxicity.PLoS Genet. 2016 Mar 29;12(3):e1005917. doi: 10.1371/journal.pgen.1005917. eCollection 2016 Mar. PLoS Genet. 2016. PMID: 27023670 Free PMC article.
-
Ca2+/calmodulin-dependent protein kinase II promotes neurodegeneration caused by tau phosphorylated at Ser262/356 in a transgenic Drosophila model of tauopathy.J Biochem. 2017 Nov 1;162(5):335-342. doi: 10.1093/jb/mvx038. J Biochem. 2017. PMID: 28992057 Free PMC article.
-
Transgenic Drosophila models of Alzheimer's disease and tauopathies.Brain Struct Funct. 2010 Mar;214(2-3):245-62. doi: 10.1007/s00429-009-0234-4. Epub 2009 Dec 5. Brain Struct Funct. 2010. PMID: 19967412 Free PMC article. Review.
-
Drosophila melanogaster as a model organism for Alzheimer's disease.Mol Neurodegener. 2013 Nov 22;8:35. doi: 10.1186/1750-1326-8-35. Mol Neurodegener. 2013. PMID: 24267573 Free PMC article. Review.
Cited by
-
Modeling the complex pathology of Alzheimer's disease in Drosophila.Exp Neurol. 2015 Dec;274(Pt A):58-71. doi: 10.1016/j.expneurol.2015.05.013. Epub 2015 May 27. Exp Neurol. 2015. PMID: 26024860 Free PMC article. Review.
-
Multiplexed Phosphoproteomic Study of Brain in Patients with Alzheimer's Disease and Age-Matched Cognitively Healthy Controls.OMICS. 2020 Apr;24(4):216-227. doi: 10.1089/omi.2019.0191. Epub 2020 Mar 17. OMICS. 2020. PMID: 32182160 Free PMC article.
-
NMNAT suppresses tau-induced neurodegeneration by promoting clearance of hyperphosphorylated tau oligomers in a Drosophila model of tauopathy.Hum Mol Genet. 2012 Jan 15;21(2):237-50. doi: 10.1093/hmg/ddr449. Epub 2011 Sep 30. Hum Mol Genet. 2012. PMID: 21965302 Free PMC article.
-
Modelling tauopathies in Drosophila: insights from the fruit fly.Int J Alzheimers Dis. 2011;2011:598157. doi: 10.4061/2011/598157. Epub 2011 Dec 29. Int J Alzheimers Dis. 2011. PMID: 22254145 Free PMC article.
-
Phenotypic differences between Drosophila Alzheimer's disease models expressing human Aβ42 in the developing eye and brain.Anim Cells Syst (Seoul). 2017 Apr 15;21(3):160-168. doi: 10.1080/19768354.2017.1313777. eCollection 2017. Anim Cells Syst (Seoul). 2017. PMID: 30460065 Free PMC article.
References
-
- Selkoe D.J. Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. - PubMed
-
- Cummings J.L. Cognitive and behavioral heterogeneity in Alzheimer's disease: seeking the neurobiological basis. Neurobiol. Aging. 2000;21:845–861. doi:10.1016/S0197-4580(00)00183-4. - DOI - PubMed
-
- Glenner G.G., Wong C.W. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984;122:1131–1135. doi:10.1016/0006-291X(84)91209-9. - DOI - PubMed
-
- Masters C.L., Simms G., Weinman N.A., Multhaup G., McDonald B.L., Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA. 1985;82:4245–4249. doi:10.1073/pnas.82.12.4245. - DOI - PMC - PubMed
-
- Sisodia S.S., St George-Hyslop P.H. Gamma-Secretase, Notch, Abeta and Alzheimer's disease: where do the presenilins fit in? Nat. Rev. Neurosci. 2002;3:281–290. doi:10.1038/nrn785. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
