

Genetic analysis of fin development in zebrafish identifies furin and hemicentin1 as potential novel fraser syndrome disease genes
- PMID: 20419147
- PMCID: PMC2855323
- DOI: 10.1371/journal.pgen.1000907
Genetic analysis of fin development in zebrafish identifies furin and hemicentin1 as potential novel fraser syndrome disease genes
Abstract
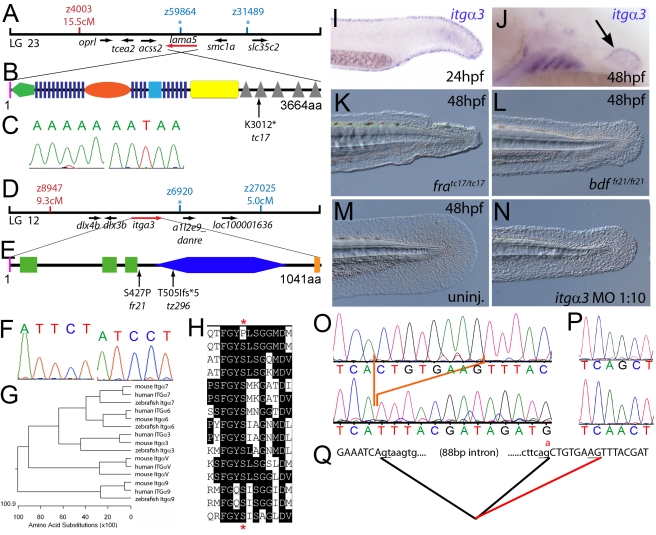
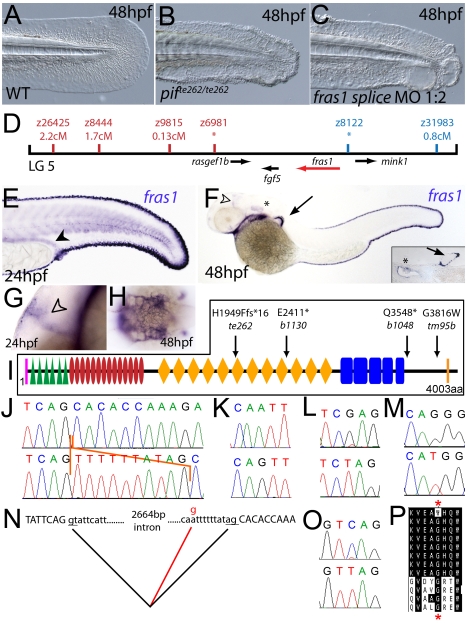
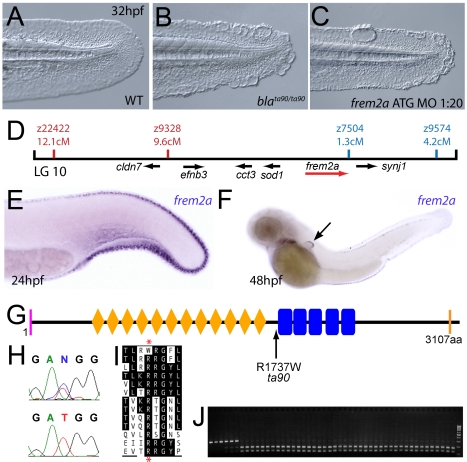
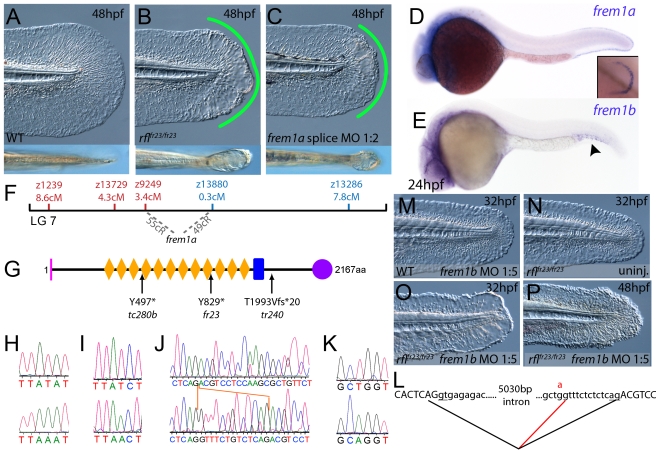
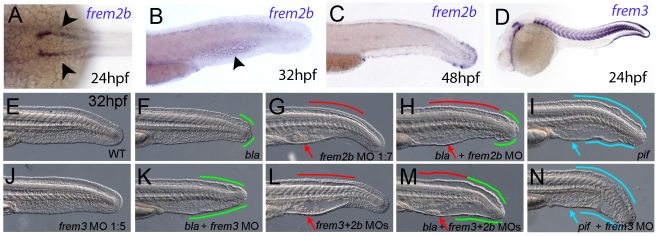
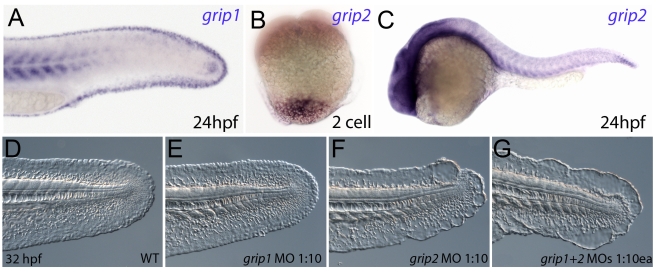
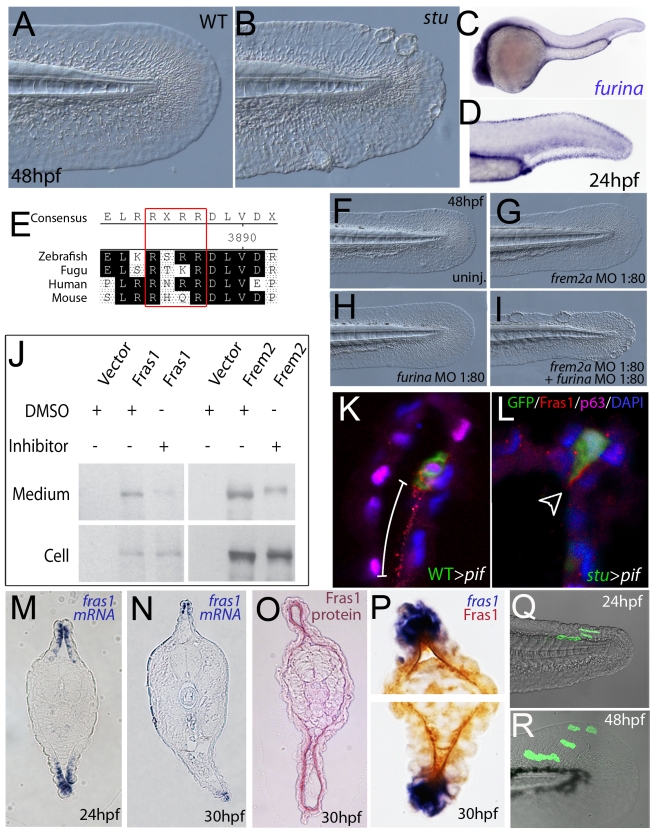
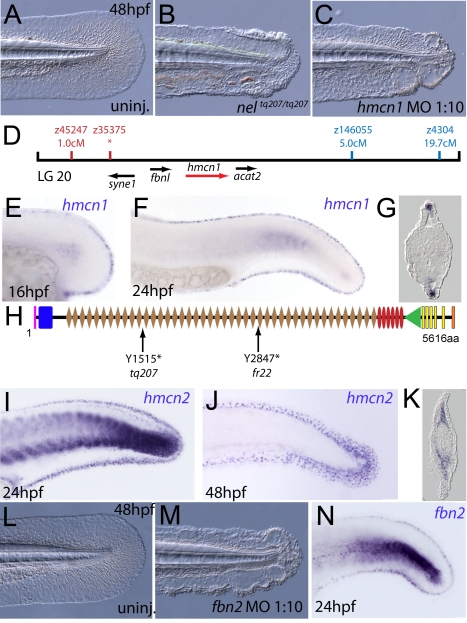
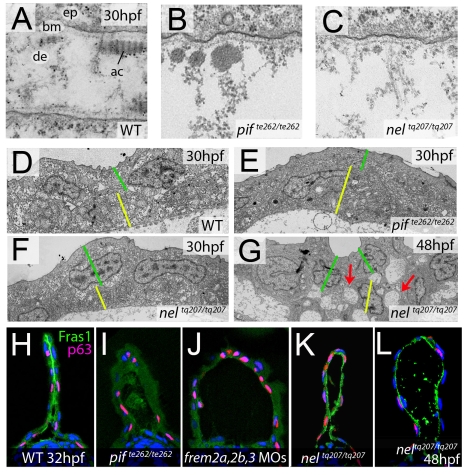
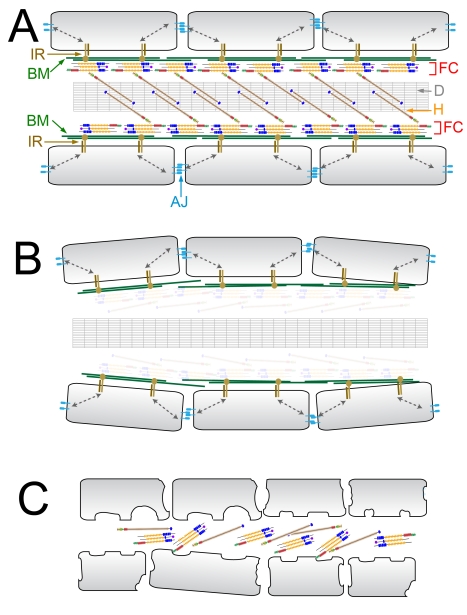
Using forward genetics, we have identified the genes mutated in two classes of zebrafish fin mutants. The mutants of the first class are characterized by defects in embryonic fin morphogenesis, which are due to mutations in a Laminin subunit or an Integrin alpha receptor, respectively. The mutants of the second class display characteristic blistering underneath the basement membrane of the fin epidermis. Three of them are due to mutations in zebrafish orthologues of FRAS1, FREM1, or FREM2, large basement membrane protein encoding genes that are mutated in mouse bleb mutants and in human patients suffering from Fraser Syndrome, a rare congenital condition characterized by syndactyly and cryptophthalmos. Fin blistering in a fourth group of zebrafish mutants is caused by mutations in Hemicentin1 (Hmcn1), another large extracellular matrix protein the function of which in vertebrates was hitherto unknown. Our mutant and dose-dependent interaction data suggest a potential involvement of Hmcn1 in Fraser complex-dependent basement membrane anchorage. Furthermore, we present biochemical and genetic data suggesting a role for the proprotein convertase FurinA in zebrafish fin development and cell surface shedding of Fras1 and Frem2, thereby allowing proper localization of the proteins within the basement membrane of forming fins. Finally, we identify the extracellular matrix protein Fibrillin2 as an indispensable interaction partner of Hmcn1. Thus we have defined a series of zebrafish mutants modelling Fraser Syndrome and have identified several implicated novel genes that might help to further elucidate the mechanisms of basement membrane anchorage and of the disease's aetiology. In addition, the novel genes might prove helpful to unravel the molecular nature of thus far unresolved cases of the human disease.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
AMACO is a component of the basement membrane-associated Fraser complex.J Invest Dermatol. 2014 May;134(5):1313-1322. doi: 10.1038/jid.2013.492. Epub 2013 Nov 14. J Invest Dermatol. 2014. PMID: 24232570 Free PMC article.
-
Breakdown of the reciprocal stabilization of QBRICK/Frem1, Fras1, and Frem2 at the basement membrane provokes Fraser syndrome-like defects.Proc Natl Acad Sci U S A. 2006 Aug 8;103(32):11981-6. doi: 10.1073/pnas.0601011103. Epub 2006 Jul 31. Proc Natl Acad Sci U S A. 2006. PMID: 16880404 Free PMC article.
-
Hemicentin 2 and Fibulin 1 are required for epidermal-dermal junction formation and fin mesenchymal cell migration during zebrafish development.Dev Biol. 2012 Sep 15;369(2):235-48. doi: 10.1016/j.ydbio.2012.06.023. Epub 2012 Jul 6. Dev Biol. 2012. PMID: 22771579 Free PMC article.
-
The role of Fras1/Frem proteins in the structure and function of basement membrane.Int J Biochem Cell Biol. 2011 Apr;43(4):487-95. doi: 10.1016/j.biocel.2010.12.016. Epub 2010 Dec 21. Int J Biochem Cell Biol. 2011. PMID: 21182980 Review.
-
The genetics of Fraser syndrome and the blebs mouse mutants.Hum Mol Genet. 2005 Oct 15;14 Spec No. 2:R269-74. doi: 10.1093/hmg/ddi262. Hum Mol Genet. 2005. PMID: 16244325 Review.
Cited by
-
A new job for ancient extracellular matrix proteins: Hemicentins stabilize cleavage furrows.Commun Integr Biol. 2011 Jul;4(4):433-5. doi: 10.4161/cib.4.4.15324. Epub 2011 Jul 1. Commun Integr Biol. 2011. PMID: 21966563 Free PMC article.
-
The role of FREM2 and FRAS1 in the development of congenital diaphragmatic hernia.Hum Mol Genet. 2018 Jun 15;27(12):2064-2075. doi: 10.1093/hmg/ddy110. Hum Mol Genet. 2018. PMID: 29618029 Free PMC article.
-
Protein-Trap Insertional Mutagenesis Uncovers New Genes Involved in Zebrafish Skin Development, Including a Neuregulin 2a-Based ErbB Signaling Pathway Required during Median Fin Fold Morphogenesis.PLoS One. 2015 Jun 25;10(6):e0130688. doi: 10.1371/journal.pone.0130688. eCollection 2015. PLoS One. 2015. PMID: 26110643 Free PMC article.
-
A basement membrane discovery pipeline uncovers network complexity, regulators, and human disease associations.Sci Adv. 2022 May 20;8(20):eabn2265. doi: 10.1126/sciadv.abn2265. Epub 2022 May 18. Sci Adv. 2022. PMID: 35584218 Free PMC article.
-
Congenital upper eyelid coloboma: embryologic, nomenclatorial, nosologic, etiologic, pathogenetic, epidemiologic, clinical, and management perspectives.Ophthalmic Plast Reconstr Surg. 2015 Jan-Feb;31(1):1-12. doi: 10.1097/IOP.0000000000000347. Ophthalmic Plast Reconstr Surg. 2015. PMID: 25419956 Free PMC article. Review.
References
-
- Jadeja S, Smyth I, Pitera JE, Taylor MS, van Haelst M, et al. Identification of a new gene mutated in Fraser syndrome and mouse myelencephalic blebs. Nat Genet. 2005;37:520–525. - PubMed
-
- McGregor L, Makela V, Darling SM, Vrontou S, Chalepakis G, et al. Fraser syndrome and mouse blebbed phenotype caused by mutations in FRAS1/Fras1 encoding a putative extracellular matrix protein. Nat Genet. 2003;34:203–208. - PubMed
-
- Vrontou S, Petrou P, Meyer BI, Galanopoulos VK, Imai K, et al. Fras1 deficiency results in cryptophthalmos, renal agenesis and blebbed phenotype in mice. Nat Genet. 2003;34:209–214. - PubMed
-
- Kiyozumi D, Sugimoto N, Nakano I, Sekiguchi K. Frem3, a member of the 12 CSPG repeats-containing extracellular matrix protein family, is a basement membrane protein with tissue distribution patterns distinct from those of Fras1, Frem2, and QBRICK/Frem1. Matrix Biol. 2007;26:456–462. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
