

Review
doi: 10.1016/j.blre.2010.03.002.
Epub 2010 Apr 24.
Pathophysiology and management of inherited bone marrow failure syndromes
Affiliations
- PMID: 20417588
- PMCID: PMC3733544
- DOI: 10.1016/j.blre.2010.03.002
Item in Clipboard
Review
Pathophysiology and management of inherited bone marrow failure syndromes
Blood Rev.
2010 May.
Erratum in
- Blood Rev. 2010 Jul-Sep;24(4-5):201
Abstract
The inherited marrow failure syndromes are a diverse set of genetic disorders characterized by hematopoietic aplasia and cancer predisposition. The clinical phenotypes are highly variable and much broader than previously recognized. The medical management of the inherited marrow failure syndromes differs from that of acquired aplastic anemia or malignancies arising in the general population. Diagnostic workup, molecular pathogenesis, and clinical treatment are reviewed.
Copyright 2010 Elsevier Ltd. All rights reserved.
Figures
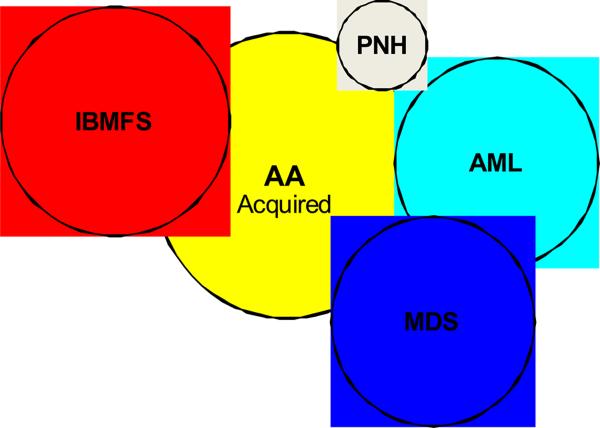
Overlapping Syndromes. The differential diagnosis for apparently acquired aplastic anemia includes paroxysmal nocturnal hemoglobinuria (PNH), myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), and inherited bone marrow failure syndromes (IBMFS).
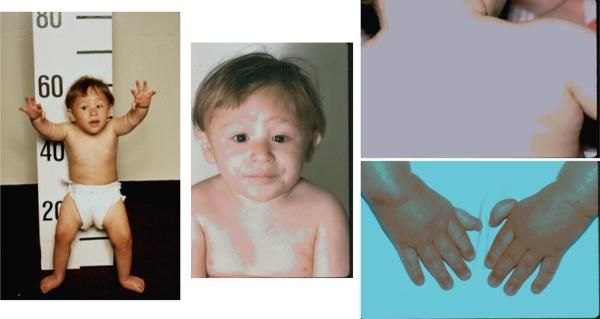
Patient with Fanconi Anemia. Features include short stature, microcephaly, dangling thumbs, epicanthal folds, micropthalmia, triangular face, café au lait and hypopigmented areas, dislocated hips which prevent him from standing straight, and rockerbottom feet. He also had an imperforate anus and ureter reimplantation. Consent for publication obtained.

Age at diagnosis of cases reported in the literature in the major IBMFS. FA, Fanconi Anemia. DC, Dyskeratosis Congenita. DBA, Diamond-Blackfan Anemia. SDS, Shwachman-Diamond Syndrome. FA, age available in 1497/2002 case reports; median age 6.5 years, range 0–49. DC, age available for 467/550 case reports; median age 14 years, range 0–75. DBA, age available for 722/980 case reports; median 3 months, range birth-64 years. SDS, age available for 318/563 case reports; median 2 weeks, range birth-11 years. Note the different X-axes. Insets in DBA and SDS extend to older patients, representing <3% of the total number for each syndrome.

Patterns of chromosomes in blood treated with DNA crosslinking agents. Left, MMC, mitomycin C, arrows show radial figures. Right, DEB, diepoxybutane, arrows show breaks, gaps, and rearrangement figures. Photograph courtesy of Lisa Moreau.
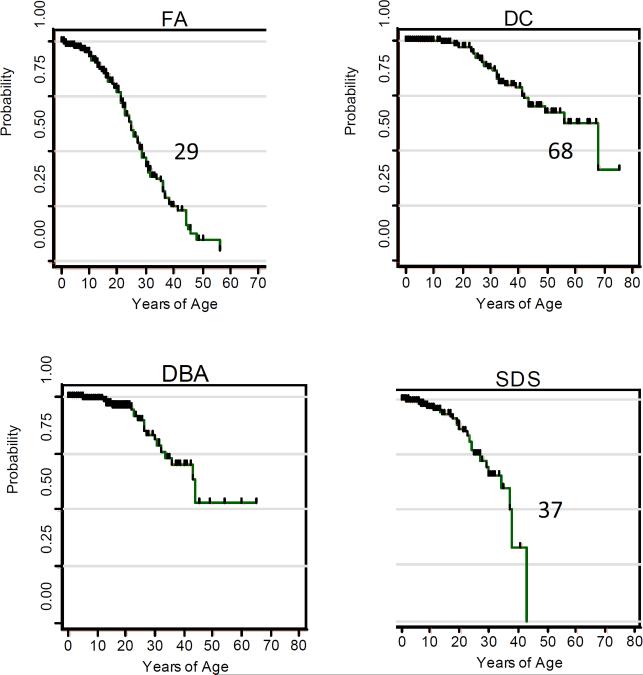
Probability of survival free of first cancer (solid tumors or leukemia) in cases reported in the literature.
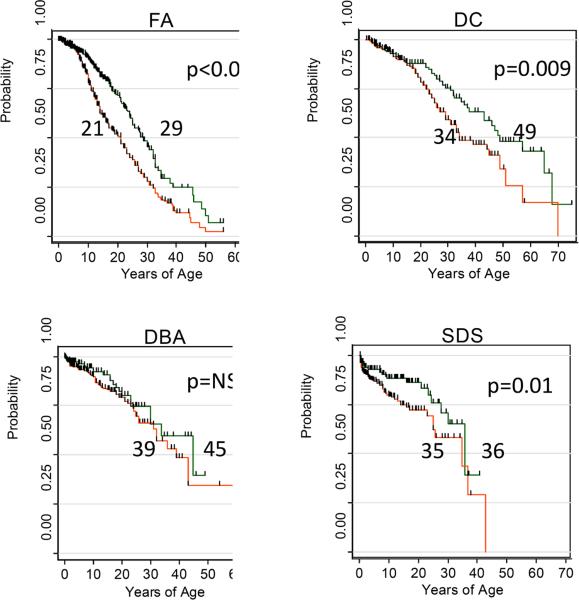
Overall survival of literature cases according to era of publication. Red, bottom line in each represents publications prior to 2000, while the green, upper line is for cases from 2000–2009. FA, median age for 1927–1999 was 21 years; 2000–2009 29 years, p <0.001. DC, median age for 1910–1999 was 34 years, 2000–2009 49 years, p = 0.009. DBA, median age for 1936–1999 was 38 years, 2000–2009 45 years, p = not significant. SDS, median age for 1949 was 35 years, 2000–2009 36 years, p = 0.01.
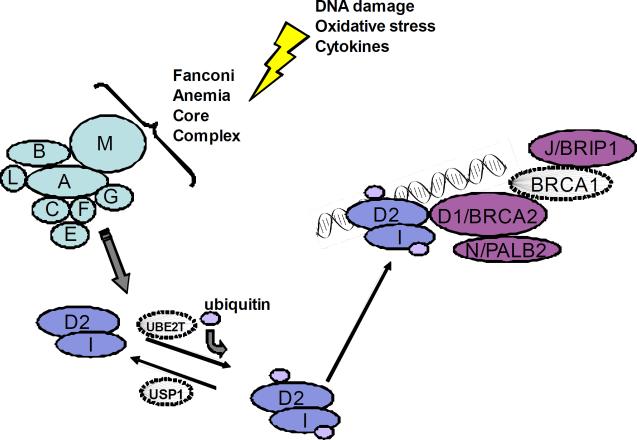
FA/BRCA DNA damage response pathway. Following DNA damage, the proteins represented by A, B, C, E, F, G, L, and M form the core complex, which is required for ubiquitination of the I and D2 proteins, which are in turn required for the downstream complex of D2-ubi, I-ubi, and D1/BRCA2, N/PALB2, BRCA1, and J/BACH1/BRIP1 to form foci for DNA repair. Only BRCA1 is not yet known to be a Fanconi gene.
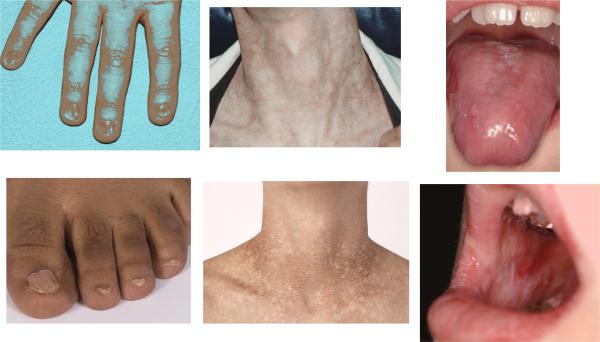
Features of the diagnostic triad in DC. Left, dystrophic nails on hands and feet. Middle, lacy reticular pigmentation on neck and upper thorax. Right, oral leukoplakia on tongue and buccal mucosa. Some of the figures are from Savage and Alter.

Cerebellar hypoplasia in the Hoyeraal-Hreidarsson variant of DC. Magnetic resonance image of brain; arrow indicates very small cerebellum.

Telomere length in blood lymphocytes according to age in patients with DC and their relatives (left), and patients with other IBMFS and their relatives (right). Vertical axis indicates telomere length in kilobases. Lines indicate the first, tenth, 50th, 90th, and 99th percentile of results from 400 normal control subjects. Left: Red circles, DC, 17 dyskeratosis congenita patients. Green triangles, HH, 4 Hoyeraal-Hreidarsson patients. Light blue diamonds, RS, 14 Revesz Syndrome patients. Dark blue square, 1 silent carrier with mutation in TERC. Open squares, 54 relatives of patients with DC. Arrows indicate 2 silent carriers initially classified as relatives, later found to have mutations in TINF2. Right: Red circles, 13 FA patients. Dark blue circle, 1 FA patient after bone marrow transplant. Light blue circles, 3 FA mosaics. Green triangle, 14 DBA patients. Black diamond, 5 SDS patients. Magenta square, 10 non-IBMFS patients. Open square, 36 relatives.
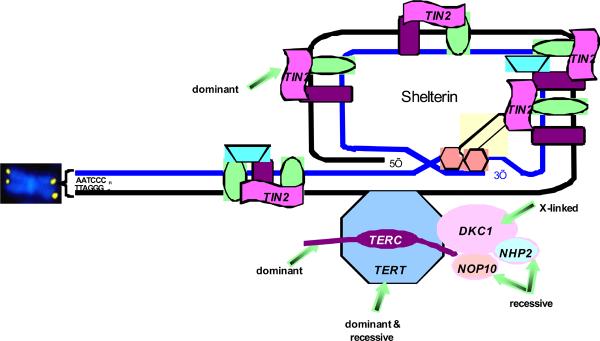
Figure 11: Telomere biology pathway with mutations in patients with DC. Telomeres are represented by the yellow dots on the ends of the chromosome (shown in blue). Telomeres are repeats of TTAGGG, added during cell replication by components of the pathway. TERT, telomerase enzyme, is a dominant. TERC, RNA template, is both dominant and recessive. DKC1, X-linked recessive gene for protein called dyskerin. NOP10/NOLA3 and NHP2/NOLA2 are autosomal recessive. TINF2 codes for the TIN2 protein, involved in maintenance of shelterin, which protects the telomere. Other proteins shown in the figure have not been found to be mutated in patients with DC. Figure courtesy of Sharon Savage.
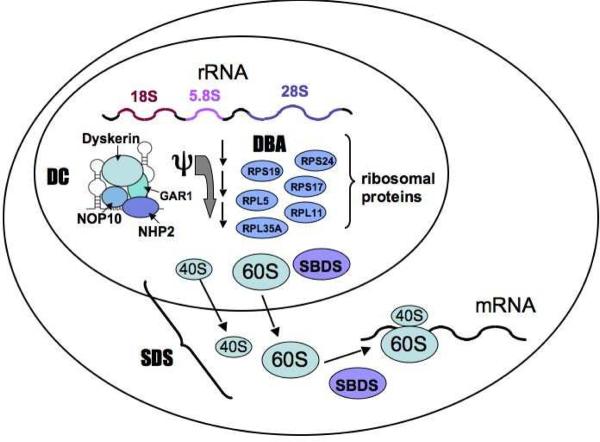
Pathways involved in ribosomal synthesis, linking DC, DBA, and SDS. The genes encoding the ribosomal protein components RPS19, RPS17, RPS24, RPL5, RPL11, and RPL35A are mutated in DBA. Mutations in these genes affect 40S and 60S ribosome biogenesis. The DKC1 gene encodes the dyskerin protein which has been implicated in ribosomal RNA pseudouridylation (ψ). The SBDS protein appears to be involved in the joining of the 40S and 60S ribosomal subunits to form the mature 80S ribosome.
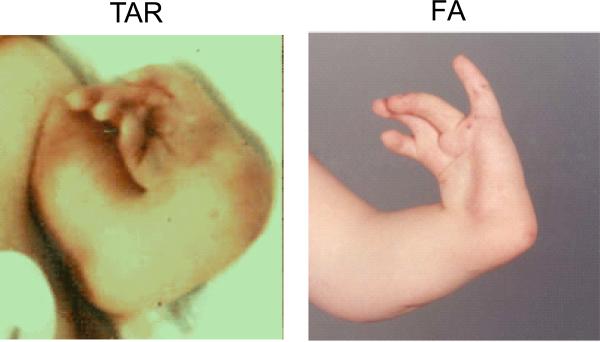
Comparison of radial ray anomalies in TAR and FA. Left, TAR. Right, FA. TAR patient has absent radii, but thumbs are present, albeit not normal in shape or position. FA patient has an absent radius, but the thumb is also absent, and the fingers are abnormal.,
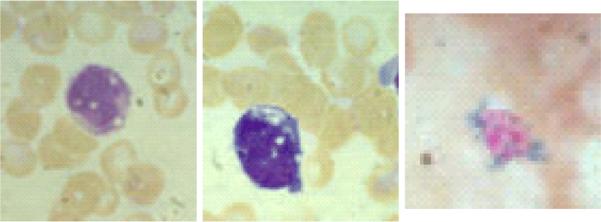
Bone marrow morphology in Pearson Syndrome. Left, vacuoles in myeloid precursor. Middle, vacuoles in erythroid precursor. Right, ringed sideroblast.
Similar articles
-
Acquired and Inherited Bone Marrow Failure Syndromes.Hematol Oncol Clin North Am. 2018 Aug;32(4):xiii-xiv. doi: 10.1016/j.hoc.2018.05.001. Hematol Oncol Clin North Am. 2018. PMID: 30047424 No abstract available.
-
Introduction to Acquired and Inherited Bone Marrow Failure.Hematol Oncol Clin North Am. 2018 Aug;32(4):569-580. doi: 10.1016/j.hoc.2018.04.008. Hematol Oncol Clin North Am. 2018. PMID: 30047411 Review.
-
Ribosomes and marrow failure: coincidental association or molecular paradigm?Blood. 2006 Jun 15;107(12):4583-8. doi: 10.1182/blood-2005-12-4831. Epub 2006 Feb 28. Blood. 2006. PMID: 16507776 Review.
-
Old and new tools in the clinical diagnosis of inherited bone marrow failure syndromes.Hematology Am Soc Hematol Educ Program. 2017 Dec 8;2017(1):79-87. doi: 10.1182/asheducation-2017.1.79. Hematology Am Soc Hematol Educ Program. 2017. PMID: 29222240 Free PMC article. Review.
-
Congenital disorders of ribosome biogenesis and bone marrow failure.Biol Blood Marrow Transplant. 2010 Jan;16(1 Suppl):S12-7. doi: 10.1016/j.bbmt.2009.09.012. Epub 2009 Sep 19. Biol Blood Marrow Transplant. 2010. PMID: 19770060 Free PMC article. Review.
Cited by
-
The use of haematopoietic stem cell transplantation in Fanconi anaemia patients: a survey of decision making among families in the US and Canada.Health Expect. 2015 Oct;18(5):929-41. doi: 10.1111/hex.12066. Epub 2013 Apr 29. Health Expect. 2015. PMID: 23621292 Free PMC article.
-
Anesthetic Management of a Patient With Fanconi Anemia.Anesth Prog. 2019 Winter;66(4):218-220. doi: 10.2344/anpr-66-02-06. Anesth Prog. 2019. PMID: 31891293 Free PMC article.
-
Non-Melanoma Skin Cancers and Other Cutaneous Manifestations in Bone Marrow Failure Syndromes and Rare DNA Repair Disorders.Front Oncol. 2022 Mar 10;12:837059. doi: 10.3389/fonc.2022.837059. eCollection 2022. Front Oncol. 2022. PMID: 35359366 Free PMC article. Review.
-
Costs and consequences of immunosuppressive therapy in children with aplastic anemia.Haematologica. 2011 Jun;96(6):793-5. doi: 10.3324/haematol.2011.044917. Haematologica. 2011. PMID: 21632841 Free PMC article. No abstract available.
-
Effective Multi-lineage Engraftment in a Mouse Model of Fanconi Anemia Using Non-genotoxic Antibody-Based Conditioning.Mol Ther Methods Clin Dev. 2020 Feb 8;17:455-464. doi: 10.1016/j.omtm.2020.02.001. eCollection 2020 Jun 12. Mol Ther Methods Clin Dev. 2020. PMID: 32226796 Free PMC article.
References
-
- Bagby GC, Meyers G. Bone marrow failure syndromes. Preface. Hematol Oncol Clin North Am. 2009;23:xiii–xiv. - PubMed
-
- Johnson MA, Olson S, Alter BP, Giri N, Hogan WJ, Richards CS. An unusual case of Fanconi Anemia with adult onset, mosaicism in an asymptomatic sibling, and a possible molecular explanation. 2009
-
- Auerbach AD, Adler B, Chaganti RS. Prenatal and postnatal diagnosis and carrier detection of Fanconi anemia by a cytogenetic method. Pediatrics. 1981;67:128–135. - PubMed
-
- Ameziane N, Errami A, Leveille F, Fontaine C, de Vries Y, van Spaendonk RM, de Winter JP, Pals G, Joenje H. Genetic subtyping of Fanconi anemia by comprehensive mutation screening. Hum Mutat. 2008;29:159–166. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
