

PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy
- PMID: 20404107
- PMCID: PMC2856912
- DOI: 10.1083/jcb.200910140
PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy
Abstract
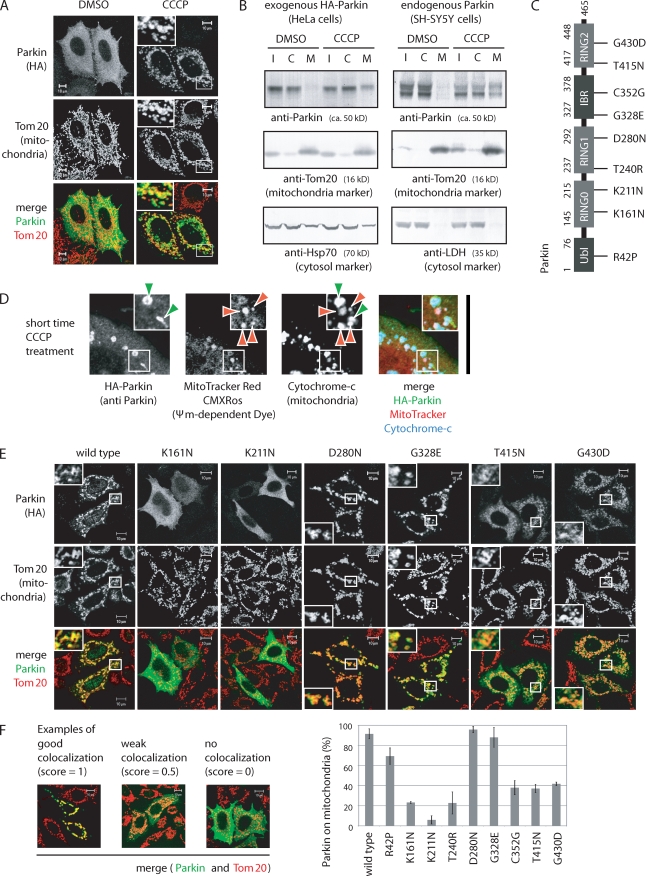
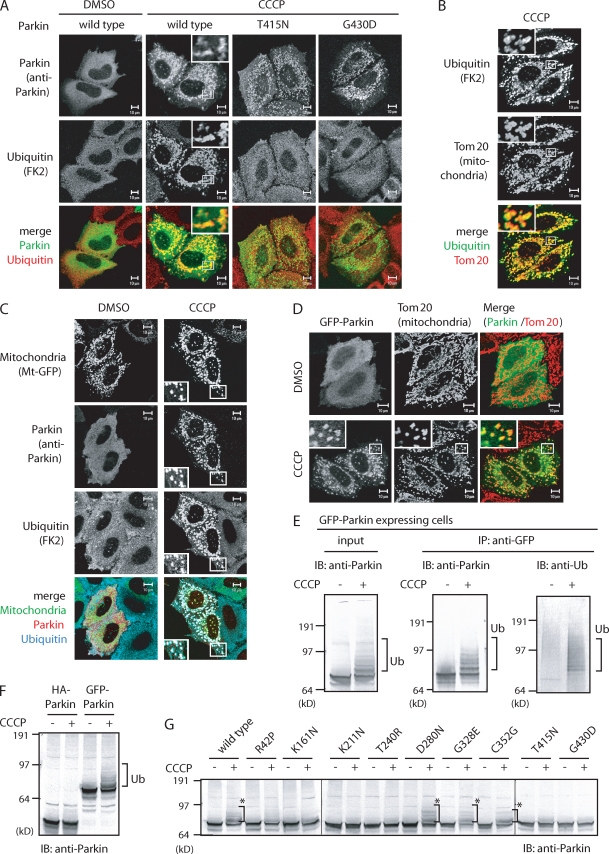
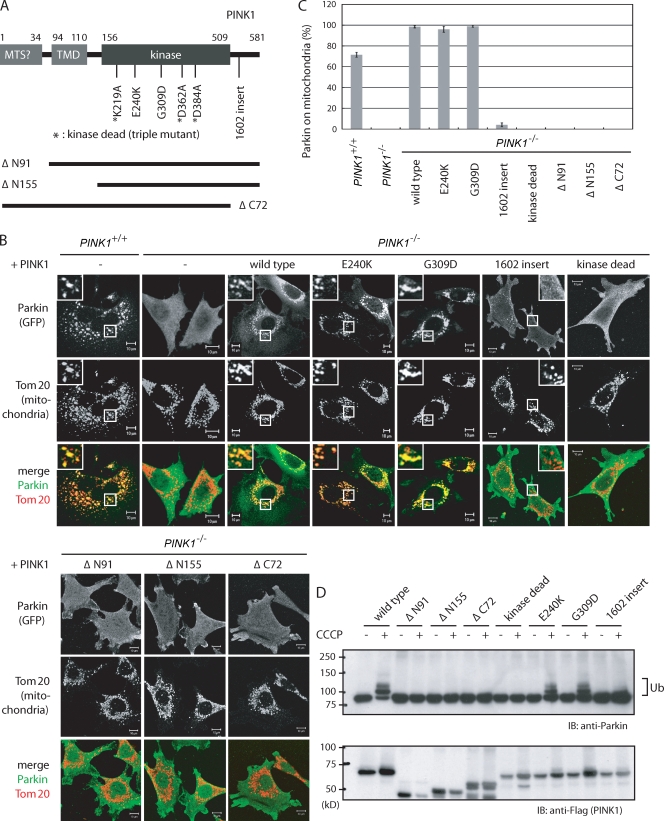
Parkinson's disease (PD) is a prevalent neurodegenerative disorder. Recent identification of genes linked to familial forms of PD such as Parkin and PINK1 (PTEN-induced putative kinase 1) has revealed that ubiquitylation and mitochondrial integrity are key factors in disease pathogenesis. However, the exact mechanism underlying the functional interplay between Parkin-catalyzed ubiquitylation and PINK1-regulated mitochondrial quality control remains an enigma. In this study, we show that PINK1 is rapidly and constitutively degraded under steady-state conditions in a mitochondrial membrane potential-dependent manner and that a loss in mitochondrial membrane potential stabilizes PINK1 mitochondrial accumulation. Furthermore, PINK1 recruits Parkin from the cytoplasm to mitochondria with low membrane potential to initiate the autophagic degradation of damaged mitochondria. Interestingly, the ubiquitin ligase activity of Parkin is repressed in the cytoplasm under steady-state conditions; however, PINK1-dependent mitochondrial localization liberates the latent enzymatic activity of Parkin. Some pathogenic mutations of PINK1 and Parkin interfere with the aforementioned events, suggesting an etiological importance. These results provide crucial insight into the pathogenic mechanisms of PD.
Figures


Similar articles
-
Mitochondrial CISD1/Cisd accumulation blocks mitophagy and genetic or pharmacological inhibition rescues neurodegenerative phenotypes in Pink1/parkin models.Mol Neurodegener. 2024 Jan 25;19(1):12. doi: 10.1186/s13024-024-00701-3. Mol Neurodegener. 2024. PMID: 38273330 Free PMC article.
-
Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin.J Cell Biol. 2010 Dec 27;191(7):1367-80. doi: 10.1083/jcb.201007013. Epub 2010 Dec 20. J Cell Biol. 2010. PMID: 21173115 Free PMC article.
-
Tom20 gates PINK1 activity and mediates its tethering of the TOM and TIM23 translocases upon mitochondrial stress.Proc Natl Acad Sci U S A. 2024 Mar 5;121(10):e2313540121. doi: 10.1073/pnas.2313540121. Epub 2024 Feb 28. Proc Natl Acad Sci U S A. 2024. PMID: 38416681 Free PMC article.
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
-
Role of AMBRA1 in mitophagy regulation: emerging evidence in aging-related diseases.Autophagy. 2024 Dec;20(12):2602-2615. doi: 10.1080/15548627.2024.2389474. Epub 2024 Sep 2. Autophagy. 2024. PMID: 39113560 Free PMC article. Review.
Cited by
-
Parkin Overexpression Attenuates Sepsis-Induced Muscle Wasting.Cells. 2020 Jun 11;9(6):1454. doi: 10.3390/cells9061454. Cells. 2020. PMID: 32545383 Free PMC article.
-
Neuronal autophagy: self-eating or self-cannibalism in Alzheimer's disease.Neurochem Res. 2013 Sep;38(9):1769-73. doi: 10.1007/s11064-013-1082-4. Epub 2013 Jun 5. Neurochem Res. 2013. PMID: 23737325 Free PMC article. Review.
-
The Organization of Mitochondrial Quality Control and Life Cycle in the Nervous System In Vivo in the Absence of PINK1.J Neurosci. 2015 Jun 24;35(25):9391-401. doi: 10.1523/JNEUROSCI.1198-15.2015. J Neurosci. 2015. PMID: 26109662 Free PMC article.
-
Vexed mutations promote degeneration of dopaminergic neurons through excessive activation of the innate immune response.NPJ Parkinsons Dis. 2022 Nov 2;8(1):147. doi: 10.1038/s41531-022-00417-5. NPJ Parkinsons Dis. 2022. PMID: 36323700 Free PMC article.
-
Activation of the E3 ubiquitin ligase Parkin.Biochem Soc Trans. 2015 Apr;43(2):269-74. doi: 10.1042/BST20140321. Biochem Soc Trans. 2015. PMID: 25849928 Free PMC article. Review.
References
-
- Beilina A., Van Der Brug M., Ahmad R., Kesavapany S., Miller D.W., Petsko G.A., Cookson M.R. 2005. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc. Natl. Acad. Sci. USA. 102:5703–5708 10.1073/pnas.0500617102 - DOI - PMC - PubMed
-
- Exner N., Treske B., Paquet D., Holmström K., Schiesling C., Gispert S., Carballo-Carbajal I., Berg D., Hoepken H.H., Gasser T., et al. 2007. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J. Neurosci. 27:12413–12418 10.1523/JNEUROSCI.0719-07.2007 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
