

Deregulation of mitochondrial membrane potential by mitochondrial insertion of granzyme B and direct Hax-1 cleavage
- PMID: 20388708
- PMCID: PMC2903387
- DOI: 10.1074/jbc.M109.086587
Deregulation of mitochondrial membrane potential by mitochondrial insertion of granzyme B and direct Hax-1 cleavage
Abstract
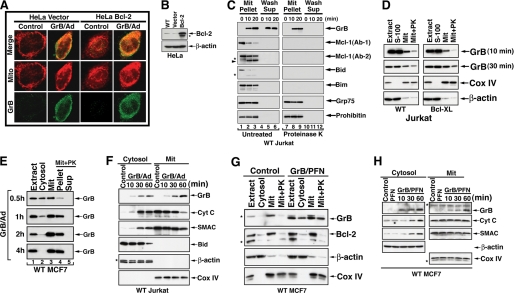
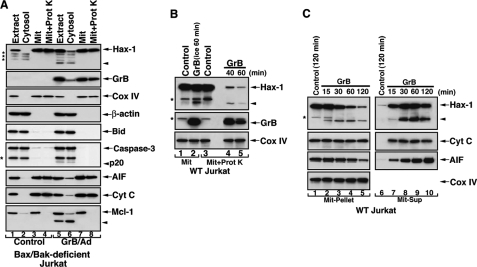
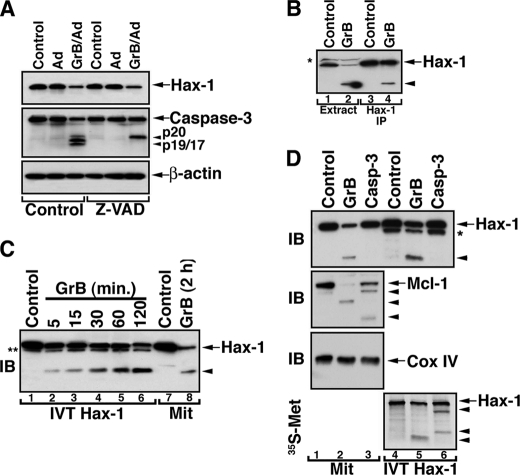
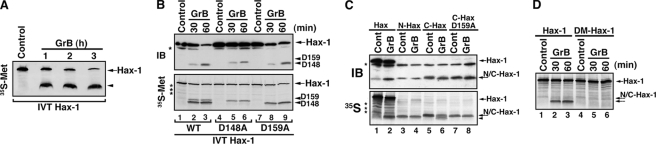
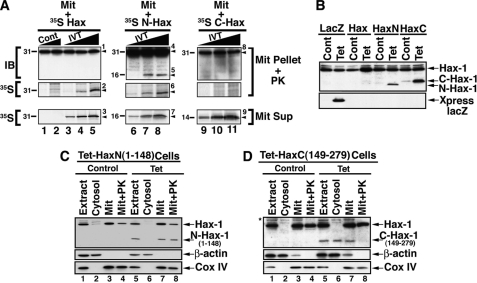
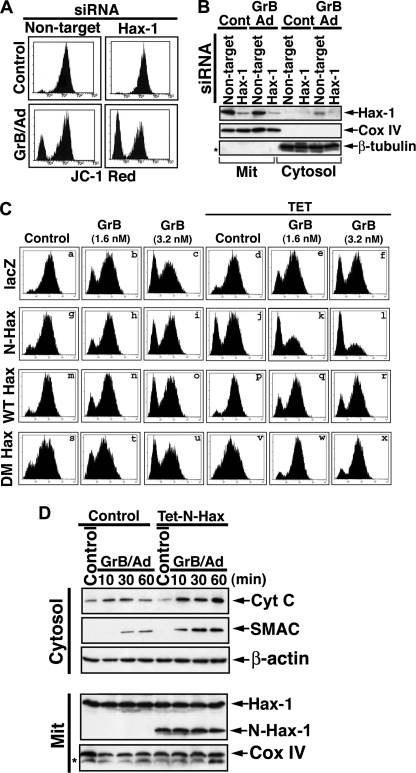
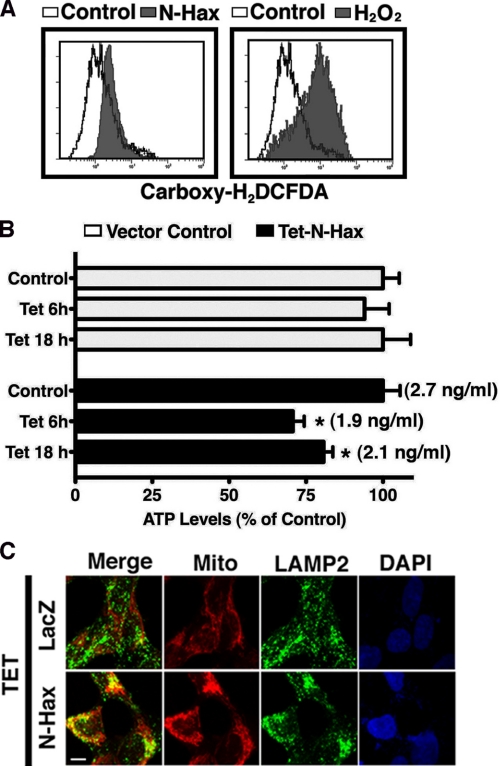
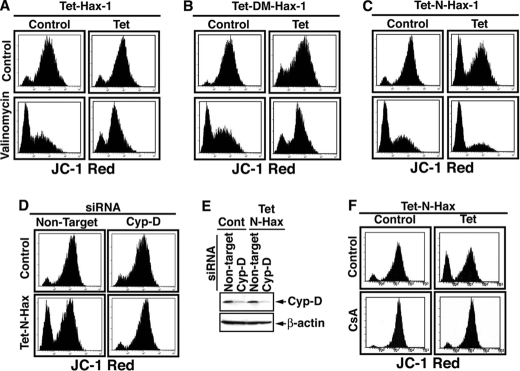
The cytoplasm and the nucleus have been identified as activity sites for granzyme B (GrB) following its delivery from cytotoxic lymphocyte granules into target cells. Here we report on the ability of exogenous GrB to insert into and function within a proteinase K-resistant mitochondrial compartment. We identified Hax-1 (HS-1-associated protein X-1), a mitochondrial protein involved in the maintenance of mitochondrial membrane potential, as a GrB substrate within the mitochondrion. GrB cleaves Hax-1 into two major fragments: an N-terminal fragment that localizes to mitochondria and a C-terminal fragment that localizes to the cytosol after being released from GrB-treated mitochondria. The N-terminal Hax-1 fragment major cellular impact is on the regulation of mitochondrial polarization. Overexpression of wild-type Hax-1 or its uncleavable mutant form protects the mitochondria against GrB or valinomycin-mediated depolarization. The N-terminal Hax-1 fragment functions as a dominant negative form of Hax-1, mediating mitochondrial depolarization in a cyclophilin D-dependent manner. Thus, induced expression of the N-terminal Hax-1 fragment results in mitochondrial depolarization and subsequent lysosomal degradation of such altered mitochondria. This study is the first to demonstrate GrB activity within the mitochondrion and to identify Hax-1 cleavage as a novel mechanism for GrB-mediated mitochondrial depolarization.
Figures
Similar articles
-
Disruption of Mcl-1.Bim complex in granzyme B-mediated mitochondrial apoptosis.J Biol Chem. 2005 Apr 22;280(16):16383-92. doi: 10.1074/jbc.M411377200. Epub 2005 Feb 15. J Biol Chem. 2005. PMID: 15713684
-
HAX-1 regulates cyclophilin-D levels and mitochondria permeability transition pore in the heart.Proc Natl Acad Sci U S A. 2015 Nov 24;112(47):E6466-75. doi: 10.1073/pnas.1508760112. Epub 2015 Nov 9. Proc Natl Acad Sci U S A. 2015. PMID: 26553996 Free PMC article.
-
Degradation of Mcl-1 by granzyme B: implications for Bim-mediated mitochondrial apoptotic events.J Biol Chem. 2004 May 21;279(21):22020-9. doi: 10.1074/jbc.M313234200. Epub 2004 Mar 10. J Biol Chem. 2004. PMID: 15014070
-
Granzyme B-induced apoptosis in cancer cells and its regulation (review).Int J Oncol. 2010 Dec;37(6):1361-78. doi: 10.3892/ijo_00000788. Int J Oncol. 2010. PMID: 21042704 Review.
-
Hax-1: a regulator of calcium signaling and apoptosis progression with multiple roles in human disease.Expert Opin Ther Targets. 2011 Jun;15(6):741-51. doi: 10.1517/14728222.2011.561787. Epub 2011 Mar 11. Expert Opin Ther Targets. 2011. PMID: 21391832 Review.
Cited by
-
Identification and characterization of OSTL (RNF217) encoding a RING-IBR-RING protein adjacent to a translocation breakpoint involving ETV6 in childhood ALL.Sci Rep. 2014 Oct 9;4:6565. doi: 10.1038/srep06565. Sci Rep. 2014. PMID: 25298122 Free PMC article.
-
4R-Cembranoid Improves Outcomes after 6-Hydroxydopamine Challenge in Both In vitro and In vivo Models of Parkinson's Disease.Front Neurosci. 2017 May 29;11:272. doi: 10.3389/fnins.2017.00272. eCollection 2017. Front Neurosci. 2017. PMID: 28611572 Free PMC article.
-
IDH2 deficiency accelerates skin pigmentation in mice via enhancing melanogenesis.Redox Biol. 2018 Jul;17:16-24. doi: 10.1016/j.redox.2018.04.008. Epub 2018 Apr 6. Redox Biol. 2018. PMID: 29660504 Free PMC article.
-
Disruption of the PRKCD-FBXO25-HAX-1 axis attenuates the apoptotic response and drives lymphomagenesis.Nat Med. 2014 Dec;20(12):1401-9. doi: 10.1038/nm.3740. Epub 2014 Nov 24. Nat Med. 2014. PMID: 25419709
-
Involvement of CASP9 (caspase 9) in IGF2R/CI-MPR endosomal transport.Autophagy. 2021 Jun;17(6):1393-1409. doi: 10.1080/15548627.2020.1761742. Epub 2020 May 25. Autophagy. 2021. PMID: 32397873 Free PMC article.
References
-
- Lieberman J., Fan Z. (2003) Curr. Opin. Immunol. 15, 553–559 - PubMed
-
- Packard B. Z., Telford W. G., Komoriya A., Henkart P. A. (2007) J. Immunol. 179, 3812–3820 - PubMed
-
- Barry M., Bleackley R. C. (2002) Nat. Rev. Immunol. 2, 401–409 - PubMed
-
- Bots M., Medema J. P. (2006) J. Cell Sci. 119, 5011–5014 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
