

PRPS1 mutations: four distinct syndromes and potential treatment
- PMID: 20380929
- PMCID: PMC2850427
- DOI: 10.1016/j.ajhg.2010.02.024
PRPS1 mutations: four distinct syndromes and potential treatment
Abstract
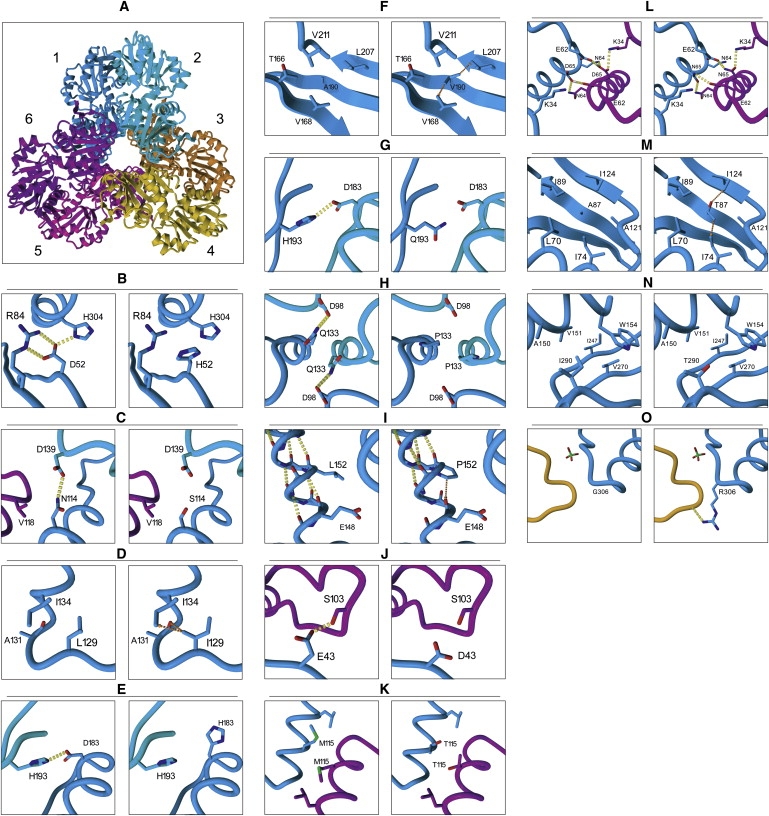
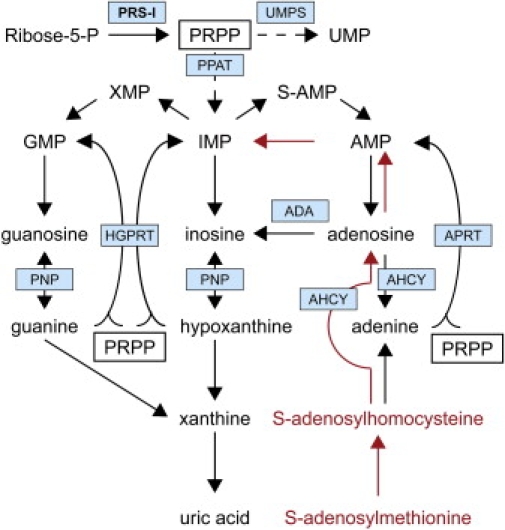
Phosphoribosylpyrophosphate synthetases (PRSs) catalyze the first step of nucleotide synthesis. Nucleotides are central to cell function, being the building blocks of nucleic acids and serving as cofactors in cellular signaling and metabolism. With this in mind, it is remarkable that mutations in phosphoribosylpyrophosphate synthetase 1 (PRPS1), which is the most ubiquitously expressed gene of the three PRS genes, are compatible with life. Mutations described thus far in PRPS1 are all missense mutations that result in PRS-I superactivity or in variable levels of decreased activity, resulting in X-linked Charcot-Marie-Tooth disease-5 (CMTX5), Arts syndrome, and X-linked nonsyndromic sensorineural deafness (DFN2). Patients with PRS-I superactivity primarily present with uric acid overproduction, mental retardation, ataxia, hypotonia, and hearing impairment. Postlingual progressive hearing loss is found as an isolated feature in DFN2 patients. Patients with CMTX5 and Arts syndrome have peripheral neuropathy, including hearing impairment and optic atrophy. However, patients with Arts syndrome are more severely affected because they also have central neuropathy and an impaired immune system. The neurological phenotype in all four PRPS1-related disorders seems to result primarily from reduced levels of GTP and possibly other purine nucleotides including ATP, suggesting that these disorders belong to the same disease spectrum. Preliminary results of S-adenosylmethionine (SAM) supplementation in two Arts syndrome patients show improvement of their condition, indicating that SAM supplementation in the diet could alleviate some of the symptoms of patients with PRPS1 spectrum diseases by replenishing purine nucleotides (J.C., unpublished data).
(c) 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Association of PRPS1 Mutations with Disease Phenotypes.Dis Markers. 2015;2015:127013. doi: 10.1155/2015/127013. Epub 2015 May 24. Dis Markers. 2015. PMID: 26089585 Free PMC article. Review.
-
Hearing loss and PRPS1 mutations: Wide spectrum of phenotypes and potential therapy.Int J Audiol. 2013 Jan;52(1):23-8. doi: 10.3109/14992027.2012.736032. Epub 2012 Nov 28. Int J Audiol. 2013. PMID: 23190330 Free PMC article. Review.
-
Expanding the phenotype of PRPS1 syndromes in females: neuropathy, hearing loss and retinopathy.Orphanet J Rare Dis. 2014 Dec 10;9:190. doi: 10.1186/s13023-014-0190-9. Orphanet J Rare Dis. 2014. PMID: 25491489 Free PMC article.
-
X-linked Charcot-Marie-Tooth disease, Arts syndrome, and prelingual non-syndromic deafness form a disease continuum: evidence from a family with a novel PRPS1 mutation.Orphanet J Rare Dis. 2014 Feb 14;9:24. doi: 10.1186/1750-1172-9-24. Orphanet J Rare Dis. 2014. PMID: 24528855 Free PMC article.
-
A Novel PRPS1 Mutation in a Japanese Patient with CMTX5.Intern Med. 2022 Jun 1;61(11):1749-1751. doi: 10.2169/internalmedicine.8029-21. Epub 2021 Nov 20. Intern Med. 2022. PMID: 34803094 Free PMC article.
Cited by
-
Analysis of miR-376 RNA cluster members in the mouse inner ear.Int J Exp Pathol. 2012 Dec;93(6):450-7. doi: 10.1111/j.1365-2613.2012.00840.x. Int J Exp Pathol. 2012. PMID: 23136997 Free PMC article.
-
PRPS2 mutations drive acute lymphoblastic leukemia relapse through influencing PRPS1/2 hexamer stability.Blood Sci. 2022 Nov 4;5(1):39-50. doi: 10.1097/BS9.0000000000000139. eCollection 2023 Jan. Blood Sci. 2022. PMID: 36742181 Free PMC article.
-
Characterizing microRNA editing and mutation sites in Autism Spectrum Disorder.Front Mol Neurosci. 2023 Jan 20;15:1105278. doi: 10.3389/fnmol.2022.1105278. eCollection 2022. Front Mol Neurosci. 2023. PMID: 36743290 Free PMC article.
-
Down-Regulation of Phosphoribosyl Pyrophosphate Synthetase 1 Inhibits Neuroblastoma Cell Proliferation.Cells. 2019 Aug 22;8(9):955. doi: 10.3390/cells8090955. Cells. 2019. PMID: 31443513 Free PMC article.
-
Nonsense mutations in SMPX, encoding a protein responsive to physical force, result in X-chromosomal hearing loss.Am J Hum Genet. 2011 May 13;88(5):621-7. doi: 10.1016/j.ajhg.2011.04.007. Epub 2011 May 5. Am J Hum Genet. 2011. PMID: 21549336 Free PMC article.
References
-
- Sperling O., Eilam G., Sara-Persky-Brosh, De Vries A. Accelerated erythrocyte 5-phosphoribosyl-1-pyrophosphate synthesis. A familial abnormality associated with excessive uric acid production and gout. Biochem. Med. 1972;6:310–316. - PubMed
-
- Kim H.J., Sohn K.M., Shy M.E., Krajewski K.M., Hwang M., Park J.H., Jang S.Y., Won H.H., Choi B.O., Hong S.H. Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (cmtx5) Am. J. Hum. Genet. 2007;81:552–558. - PMC - PubMed
-
- Rosenberg R.N., Chutorian A. Familial opticoacoustic nerve degeneration and polyneuropathy. Neurology. 1967;17:827–832. - PubMed
-
- Arts W.F., Loonen M.C., Sengers R.C., Slooff J.L. X-linked ataxia, weakness, deafness, and loss of vision in early childhood with a fatal course. Ann. Neurol. 1993;33:535–539. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
