

Shared mechanisms for opioid tolerance and a transition to chronic pain
- PMID: 20357116
- PMCID: PMC2857996
- DOI: 10.1523/JNEUROSCI.5530-09.2010
Shared mechanisms for opioid tolerance and a transition to chronic pain
Abstract
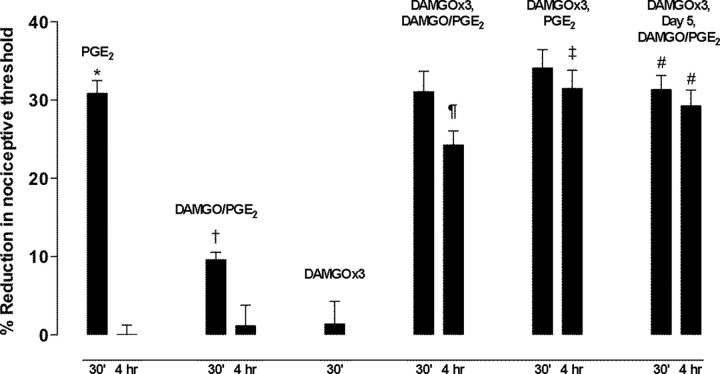
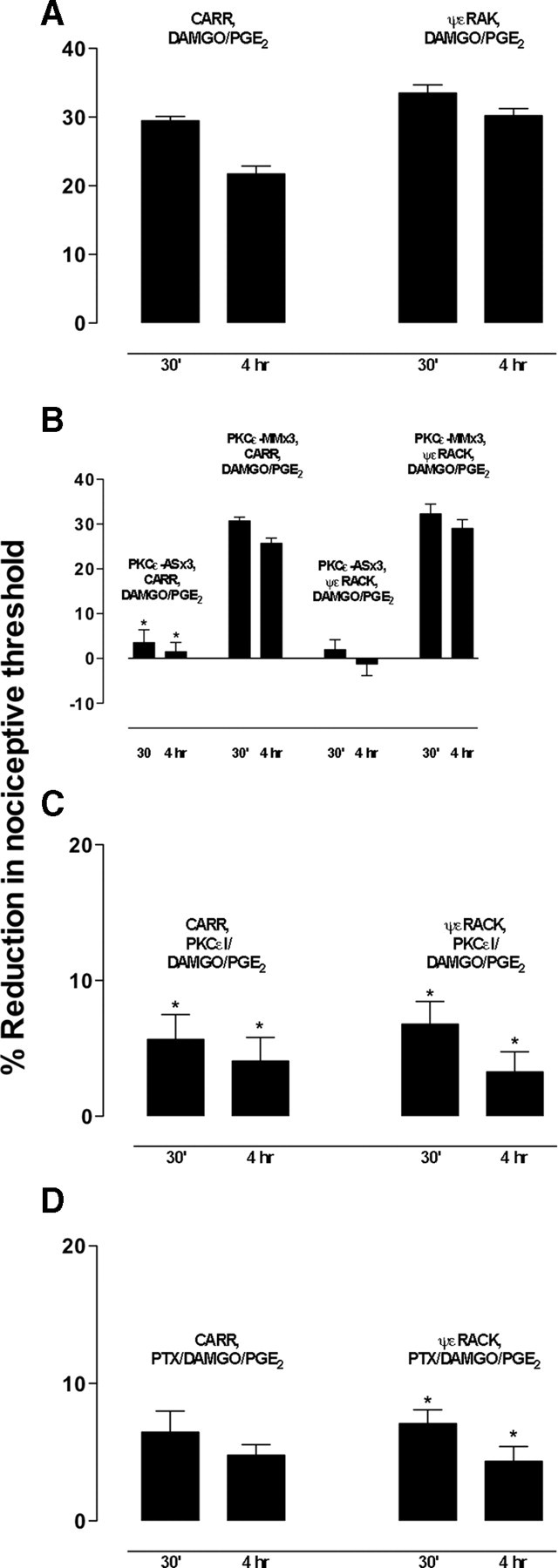
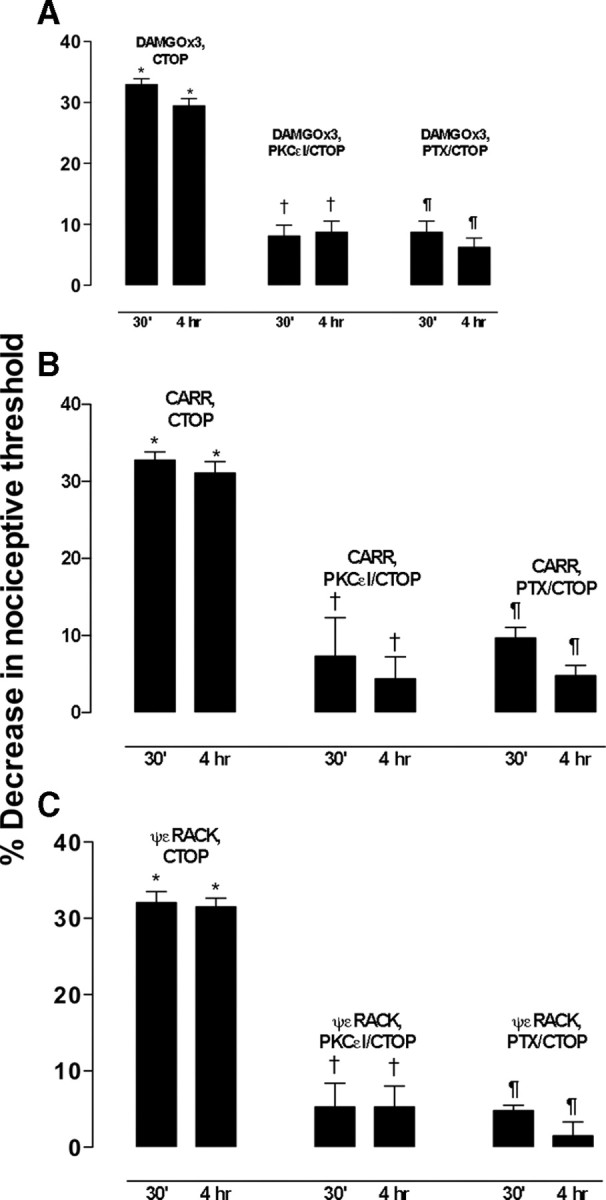
Clinical pain conditions may remain responsive to opiate analgesics for extended periods, but such persistent acute pain can undergo a transition to an opiate-resistant chronic pain state that becomes a much more serious clinical problem. To test the hypothesis that cellular mechanisms of chronic pain in the primary afferent also contribute to the development of opiate resistance, we used a recently developed model of the transition of from acute to chronic pain, hyperalgesic priming. Repeated intradermal administration of the potent and highly selective mu-opioid agonist, [d-Ala(2),N-MePhe(4),gly-ol]-enkephalin (DAMGO), to produce tolerance for its inhibition of prostaglandin E(2) hyperalgesia, simultaneously produced hyperalgesic priming. Conversely, injection of an inflammogen, carrageenan, used to produce priming produced DAMGO tolerance. Both effects were prevented by inhibition of protein kinase Cepsilon (PKCepsilon). Carrageenan also induced opioid dependence, manifest as mu-opioid receptor antagonist (d-Phe-Cys-Tyr-d-Trp-Orn-Thr-Pen-Thr-NH(2))-induced hyperalgesia that, like priming, was PKCepsilon and G(i) dependent. These findings suggest that the transition from acute to chronic pain, and development of mu-opioid receptor tolerance and dependence may be linked by common cellular mechanisms in the primary afferent.
Figures


Similar articles
-
Repeated Mu-Opioid Exposure Induces a Novel Form of the Hyperalgesic Priming Model for Transition to Chronic Pain.J Neurosci. 2015 Sep 9;35(36):12502-17. doi: 10.1523/JNEUROSCI.1673-15.2015. J Neurosci. 2015. PMID: 26354917 Free PMC article.
-
Multiple receptors involved in peripheral alpha 2, mu, and A1 antinociception, tolerance, and withdrawal.J Neurosci. 1997 Jan 15;17(2):735-44. doi: 10.1523/JNEUROSCI.17-02-00735.1997. J Neurosci. 1997. PMID: 8987795 Free PMC article.
-
Opioid-Induced Pronociceptive Signaling in the Gastrointestinal Tract Is Mediated by Delta-Opioid Receptor Signaling.J Neurosci. 2022 Apr 20;42(16):3316-3328. doi: 10.1523/JNEUROSCI.2098-21.2022. Epub 2022 Mar 7. J Neurosci. 2022. PMID: 35256532 Free PMC article.
-
Critical role of nociceptor plasticity in chronic pain.Trends Neurosci. 2009 Dec;32(12):611-8. doi: 10.1016/j.tins.2009.07.007. Epub 2009 Sep 24. Trends Neurosci. 2009. PMID: 19781793 Free PMC article. Review.
-
Imaging opioid analgesia in the human brain and its potential relevance for understanding opioid use in chronic pain.Neuropharmacology. 2014 Sep;84(100):123-30. doi: 10.1016/j.neuropharm.2013.06.035. Epub 2013 Jul 25. Neuropharmacology. 2014. PMID: 23891639 Free PMC article. Review.
Cited by
-
Transient decrease in nociceptor GRK2 expression produces long-term enhancement in inflammatory pain.Neuroscience. 2012 Oct 11;222:392-403. doi: 10.1016/j.neuroscience.2012.07.004. Epub 2012 Jul 13. Neuroscience. 2012. PMID: 22796071 Free PMC article.
-
Opioid Analgesics and Persistent Pain After an Acute Pain Emergency Department Visit: Evidence from a Cohort of Suspected Urolithiasis Patients.J Emerg Med. 2021 Dec;61(6):637-648. doi: 10.1016/j.jemermed.2021.09.002. Epub 2021 Oct 21. J Emerg Med. 2021. PMID: 34690022 Free PMC article.
-
Females Are More Likely Than Males to Fill an Opioid Prescription in the Year After Anterior Cruciate Ligament Reconstruction.Arthrosc Sports Med Rehabil. 2023 Jul 8;5(4):100758. doi: 10.1016/j.asmr.2023.100758. eCollection 2023 Aug. Arthrosc Sports Med Rehabil. 2023. PMID: 37645396 Free PMC article.
-
Acidosis Mediates the Switching of Gs-PKA and Gi-PKCε Dependence in Prolonged Hyperalgesia Induced by Inflammation.PLoS One. 2015 May 1;10(5):e0125022. doi: 10.1371/journal.pone.0125022. eCollection 2015. PLoS One. 2015. PMID: 25933021 Free PMC article.
-
Mu-opioid receptor and receptor tyrosine kinase crosstalk: Implications in mechanisms of opioid tolerance, reduced analgesia to neuropathic pain, dependence, and reward.Front Syst Neurosci. 2022 Dec 1;16:1059089. doi: 10.3389/fnsys.2022.1059089. eCollection 2022. Front Syst Neurosci. 2022. PMID: 36532632 Free PMC article. Review.
References
-
- Bailey CP, Kelly E, Henderson G. Protein kinase C activation enhances morphine-induced rapid desensitization of mu-opioid receptors in mature rat locus ceruleus neurons. Mol Pharmacol. 2004;66:1592–1598. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials