

Human cytomegalovirus protein pUL117 targets the mini-chromosome maintenance complex and suppresses cellular DNA synthesis
- PMID: 20333247
- PMCID: PMC2841624
- DOI: 10.1371/journal.ppat.1000814
Human cytomegalovirus protein pUL117 targets the mini-chromosome maintenance complex and suppresses cellular DNA synthesis
Abstract
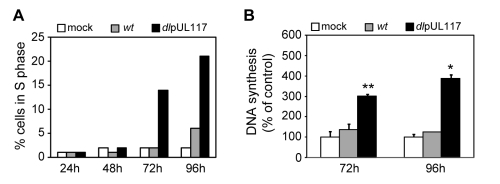
Modulation of host DNA synthesis is essential for many viruses to establish productive infections and contributes to viral diseases. Human cytomegalovirus (HCMV), a large DNA virus, blocks host DNA synthesis and deregulates cell cycle progression. We report that pUL117, a viral protein that we recently identified, is required for HCMV to block host DNA synthesis. Mutant viruses in which pUL117 was disrupted, either by frame-shift mutation or by a protein destabilization-based approach, failed to block host DNA synthesis at times after 24 hours post infection in human foreskin fibroblasts. Furthermore, pUL117-deficient virus stimulated quiescent fibroblasts to enter S-phase, demonstrating the intrinsic ability of HCMV to promote host DNA synthesis, which was suppressed by pUL117. We examined key proteins known to be involved in inhibition of host DNA synthesis in HCMV infection, and found that many were unlikely involved in the inhibitory activity of pUL117, including geminin, cyclin A, and viral protein IE2, based on their expression patterns. However, the ability of HCMV to delay the accumulation of the mini-chromosome maintenance (MCM) complex proteins, represented by MCM2 and MCM4, and prevent their loading onto chromatin, was compromised in the absence of pUL117. When expressed alone, pUL117 slowed cell proliferation, delayed DNA synthesis, and inhibited MCM accumulation. Knockdown of MCM proteins by siRNA restored the ability of pUL117-deficient virus to block cellular DNA synthesis. Thus, targeting MCM complex is one mechanism pUL117 employs to help block cellular DNA synthesis during HCMV infection. Our finding substantiates an emerging picture that deregulation of MCM is a conserved strategy for many viruses to prevent host DNA synthesis and helps to elucidate the complex strategy used by a large DNA virus to modulate cellular processes to promote infection and pathogenesis.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures








Similar articles
-
Human cytomegalovirus prevents replication licensing by inhibiting MCM loading onto chromatin.EMBO Rep. 2003 Jan;4(1):42-6. doi: 10.1038/sj.embor.embor707. EMBO Rep. 2003. PMID: 12524519 Free PMC article.
-
The full-length protein encoded by human cytomegalovirus gene UL117 is required for the proper maturation of viral replication compartments.J Virol. 2008 Apr;82(7):3452-65. doi: 10.1128/JVI.01964-07. Epub 2008 Jan 23. J Virol. 2008. PMID: 18216115 Free PMC article.
-
Human cytomegalovirus infection leads to accumulation of geminin and inhibition of the licensing of cellular DNA replication.J Virol. 2003 Feb;77(4):2369-76. doi: 10.1128/jvi.77.4.2369-2376.2003. J Virol. 2003. PMID: 12551974 Free PMC article.
-
Human cytomegalovirus riding the cell cycle.Med Microbiol Immunol. 2015 Jun;204(3):409-19. doi: 10.1007/s00430-015-0396-z. Epub 2015 Mar 17. Med Microbiol Immunol. 2015. PMID: 25776080 Review.
-
Intrinsic Immune Mechanisms Restricting Human Cytomegalovirus Replication.Viruses. 2021 Jan 26;13(2):179. doi: 10.3390/v13020179. Viruses. 2021. PMID: 33530304 Free PMC article. Review.
Cited by
-
Human cytomegalovirus pUL97 kinase induces global changes in the infected cell phosphoproteome.Proteomics. 2015 Jun;15(12):2006-22. doi: 10.1002/pmic.201400607. Epub 2015 May 12. Proteomics. 2015. PMID: 25867546 Free PMC article.
-
G1/S Cell Cycle Induction by Epstein-Barr Virus BORF2 Is Mediated by P53 and APOBEC3B.J Virol. 2022 Sep 28;96(18):e0066022. doi: 10.1128/jvi.00660-22. Epub 2022 Sep 7. J Virol. 2022. PMID: 36069545 Free PMC article.
-
Human cytomegalovirus: bacterial artificial chromosome (BAC) cloning and genetic manipulation.Curr Protoc Microbiol. 2012 Feb;Chapter 14:Unit14E.4. doi: 10.1002/9780471729259.mc14e04s24. Curr Protoc Microbiol. 2012. PMID: 22307551 Free PMC article.
-
An intein-mediated modulation of protein stability system and its application to study human cytomegalovirus essential gene function.Sci Rep. 2016 May 18;6:26167. doi: 10.1038/srep26167. Sci Rep. 2016. PMID: 27188239 Free PMC article.
-
The Cytomegalovirus Tegument Protein UL35 Antagonizes Pattern Recognition Receptor-Mediated Type I IFN Transcription.Microorganisms. 2020 May 26;8(6):790. doi: 10.3390/microorganisms8060790. Microorganisms. 2020. PMID: 32466380 Free PMC article.
References
-
- Hume AJ, Finkel JS, Kamil JP, Coen DM, Culbertson MR, et al. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science. 2008;320:797–799. - PubMed
-
- Helt AM, Galloway DA. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis. 2003;24:159–169. - PubMed
-
- Mocarski ES, Shenk T, Pass RF, editors. Cytomegaloviruses. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 2701–2772.
-
- Sanchez V, Spector DH. Subversion of cell cycle regulatory pathways. Curr Top Microbiol Immunol. 2008;325:243–262. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
