

Ventilator-induced inflammatory response in lipopolysaccharide-exposed rat lung is mediated by angiotensin-converting enzyme
- PMID: 20304959
- PMCID: PMC2861087
- DOI: 10.2353/ajpath.2010.090565
Ventilator-induced inflammatory response in lipopolysaccharide-exposed rat lung is mediated by angiotensin-converting enzyme
Abstract
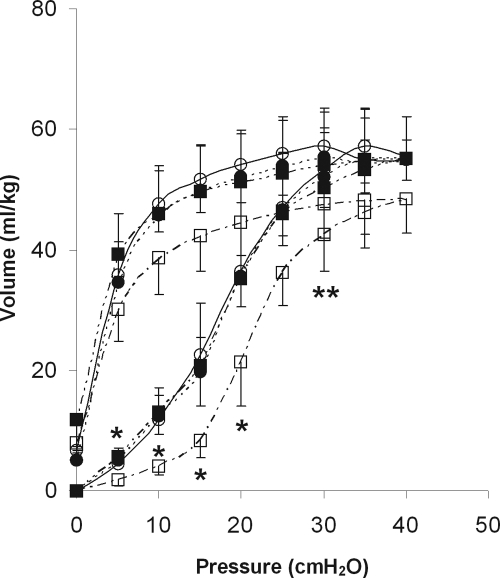
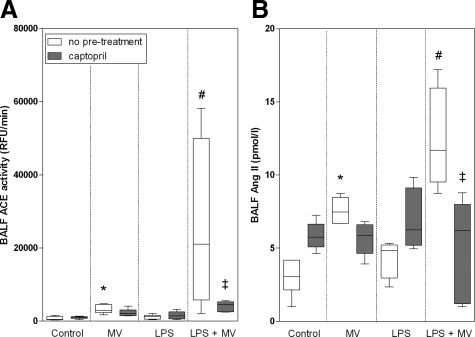
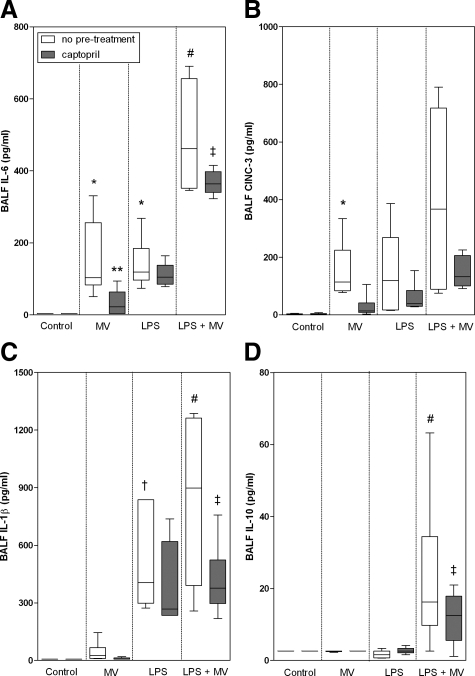
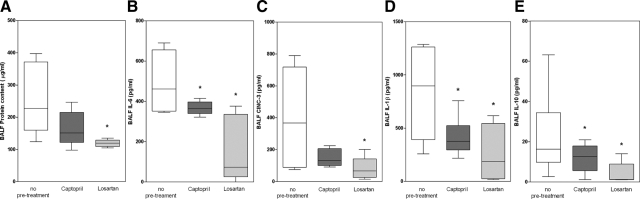
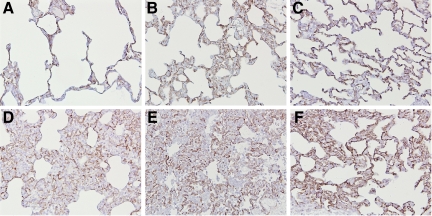
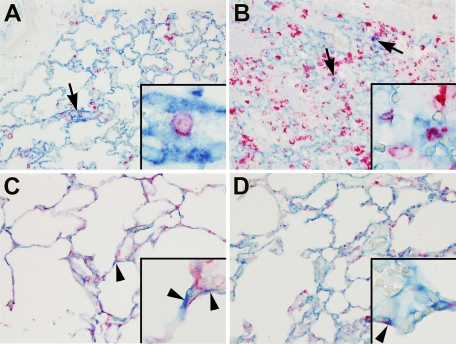
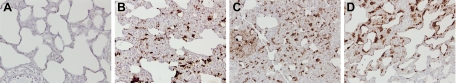
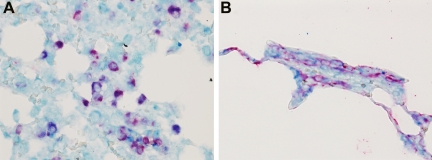
Angiotensin-converting enzyme (ACE) mediates the ventilator-induced inflammatory response in healthy lungs via angiotensin II (Ang II). A rat model was used to examine the role of ACE and Ang II in the inflammatory response during mechanical ventilation of preinjured (ie, lipopolysaccharide [LPS]-exposed) lungs. When indicated, rats were pretreated with the ACE inhibitor captopril and/or intratracheal administration of LPS. The animals were ventilated for 4 hours with moderate pressure amplitudes. Nonventilated animals served as controls. ACE activity and levels of Ang II and inflammatory mediators (interleukin-6, Cytokine-induced Neutrophil Chemoattractant (CINC)-3, interleukin-1beta, and interleukin-10) were determined in bronchoalveolar lavage fluid (BALF). The localization of ACE and Ang II type 1 receptor in lung tissue was determined by immunohistochemistry. The role of the Ang II pathway was assessed by using its receptor antagonist Losartan. Mechanical ventilation of LPS-exposed animals increased ACE activity and levels of inflammatory mediators in BALF compared with ventilated nonexposed and LPS-exposed nonventilated animals. Blocking ACE by captopril attenuated the lung inflammatory response. Furthermore, increased ACE activity in BALF was accompanied by increased levels of Ang II and enhanced expression of its receptor on alveolar cells. Blocking the Ang II receptor attenuated the inflammatory mediator response to a larger extent than by blocking ACE. In conclusion, during mechanical ventilation ACE, via Ang II, mediates the inflammatory response of both healthy and preinjured lungs.
Figures

Similar articles
-
ACE mediates ventilator-induced lung injury in rats via angiotensin II but not bradykinin.Eur Respir J. 2008 Feb;31(2):363-71. doi: 10.1183/09031936.00060207. Epub 2007 Oct 24. Eur Respir J. 2008. PMID: 17959639
-
Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist.J Pathol. 2011 Dec;225(4):618-27. doi: 10.1002/path.2987. Epub 2011 Oct 18. J Pathol. 2011. PMID: 22009550
-
Angiotensin-converting enzyme inhibition attenuates lipopolysaccharide-induced lung injury by regulating the balance between angiotensin-converting enzyme and angiotensin-converting enzyme 2 and inhibiting mitogen-activated protein kinase activation.Shock. 2015 Apr;43(4):395-404. doi: 10.1097/SHK.0000000000000302. Shock. 2015. PMID: 25768373
-
Role of the renin-angiotensin system in ventilator-induced lung injury: an in vivo study in a rat model.Thorax. 2007 Jun;62(6):527-35. doi: 10.1136/thx.2006.061945. Epub 2007 Jan 18. Thorax. 2007. PMID: 17234658 Free PMC article.
-
Losartan attenuates ventilator-induced lung injury.J Surg Res. 2008 Mar;145(1):25-32. doi: 10.1016/j.jss.2007.03.075. Epub 2007 Aug 3. J Surg Res. 2008. PMID: 17688881
Cited by
-
Classic and Nonclassic Renin-Angiotensin Systems in the Critically Ill.Crit Care Clin. 2019 Apr;35(2):213-227. doi: 10.1016/j.ccc.2018.11.002. Epub 2019 Jan 28. Crit Care Clin. 2019. PMID: 30784605 Free PMC article. Review.
-
Angiotensin-converting enzyme inhibitors (ACEI) or angiotensin receptor blockers (ARBs) may be safe for COVID-19 patients.BMC Infect Dis. 2021 Jan 25;21(1):114. doi: 10.1186/s12879-021-05821-5. BMC Infect Dis. 2021. PMID: 33494713 Free PMC article.
-
Nicotinamide exacerbates hypoxemia in ventilator-induced lung injury independent of neutrophil infiltration.PLoS One. 2015 Apr 13;10(4):e0123460. doi: 10.1371/journal.pone.0123460. eCollection 2015. PLoS One. 2015. PMID: 25875775 Free PMC article.
-
Ventilation following established ARDS: a preclinical model framework to improve predictive power.Thorax. 2019 Dec;74(12):1120-1129. doi: 10.1136/thoraxjnl-2019-213460. Epub 2019 Jul 5. Thorax. 2019. PMID: 31278170 Free PMC article.
-
Autophagy Protects Against Developing Increased Lung Permeability and Hypoxemia by Down Regulating Inflammasome Activity and IL-1β in LPS Plus Mechanical Ventilation-Induced Acute Lung Injury.Front Immunol. 2020 Feb 14;11:207. doi: 10.3389/fimmu.2020.00207. eCollection 2020. Front Immunol. 2020. PMID: 32117318 Free PMC article.
References
-
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. - PubMed
-
- Dreyfuss D, Saumon G. Ventilator-induced lung injury: lessons from experimental studies. Am J Resp Crit Care Med. 1998;157:294–323. - PubMed
-
- Tremblay LN, Slutsky AS. Ventilator-induced lung injury: from barotrauma to biotrauma. Proc Assoc Am Physicians. 1998;110:482–488. - PubMed
-
- Altemeier WA, Matute-Bello G, Gharib SA, Glenny RW, Martin TR, Liles WC. Modulation of lipopolysaccharide-induced gene transcription and promotion of lung injury by mechanical ventilation. J Immunol. 2005;175:3369–3376. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
