

THAP1 mutations (DYT6) are an additional cause of early-onset dystonia
- PMID: 20211909
- PMCID: PMC2839194
- DOI: 10.1212/WNL.0b013e3181d5276d
THAP1 mutations (DYT6) are an additional cause of early-onset dystonia
Abstract
Background: The clinical phenotype of DYT6 consists mainly of primary craniocervical dystonia. Recently, the THAP1 gene was identified as the cause of DYT6, where a total of 13 mutations have been identified in Amish-Mennonite and European families.
Methods: We sequenced the THAP1 gene in a series of 362 British, genetically undetermined, primary dystonia patients (78 with focal, 186 with segmental, and 98 with generalized dystonia) and in 28 dystonia-manifesting DYT1 patients and 176 normal control individuals.
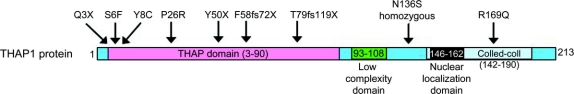
Results: Nine coding mutations were identified in the THAP1 gene. Two were small deletions, 2 were nonsense, and 5 were missense. Eight mutations were heterozygous, and 1 was homozygous. The main clinical presentation of cases with THAP1 mutations was early-onset (<30 years) dystonia in the craniocervical region or the limbs (8 of 9 patients). There was phenotypic variability with laryngeal or oromandibular dystonia present in 3 cases. Four of 9 THAP1 cases developed generalized dystonia.
Conclusions: The number of THAP1 mutations has been significantly expanded, indicating an uncommon but important cause of dystonia. Coding mutations account for 9 of 362 dystonia cases, indicating a mutation frequency of 2.5% of dystonia cases in the population that we have screened. The majority of cases reported here with THAP1 mutations had craniocervical- or limb-onset segmental dystonia, but we also identified 1 homozygous THAP1 mutation, associated initially with writer's dystonia and then developing segmental dystonia. Three of our patients had a nonsense or frameshift THAP1 mutation and the clinical features of laryngeal or oromandibular dystonia. These data suggest that early-onset dystonia that includes the involvement of the larynx or face is frequently associated with THAP1 mutations.
Figures
Similar articles
-
Genotype-phenotype correlations in THAP1 dystonia: molecular foundations and description of new cases.Parkinsonism Relat Disord. 2012 Jun;18(5):414-25. doi: 10.1016/j.parkreldis.2012.02.001. Epub 2012 Feb 28. Parkinsonism Relat Disord. 2012. PMID: 22377579 Free PMC article. Review.
-
Mutations in THAP1 (DYT6) in early-onset dystonia: a genetic screening study.Lancet Neurol. 2009 May;8(5):441-6. doi: 10.1016/S1474-4422(09)70081-X. Epub 2009 Apr 1. Lancet Neurol. 2009. PMID: 19345147 Free PMC article.
-
Mutations in THAP1 (DYT6) and generalised dystonia with prominent spasmodic dysphonia: a genetic screening study.Lancet Neurol. 2009 May;8(5):447-52. doi: 10.1016/S1474-4422(09)70083-3. Epub 2009 Apr 1. Lancet Neurol. 2009. PMID: 19345148
-
Clinical neuroimaging and electrophysiological assessment of three DYT6 dystonia families.Mov Disord. 2010 Oct 30;25(14):2405-12. doi: 10.1002/mds.23279. Mov Disord. 2010. PMID: 20687193
-
THAP1 mutations and dystonia phenotypes: genotype phenotype correlations.Mov Disord. 2012 Sep 1;27(10):1290-4. doi: 10.1002/mds.25146. Epub 2012 Aug 17. Mov Disord. 2012. PMID: 22903657 Free PMC article. Review.
Cited by
-
Mutation screening of the DYT6/THAP1 gene in Serbian patients with primary dystonia.J Neurol. 2013 Apr;260(4):1037-42. doi: 10.1007/s00415-012-6753-6. Epub 2012 Nov 20. J Neurol. 2013. PMID: 23180184
-
Advances in the genetics of primary torsion dystonia.F1000 Biol Rep. 2010 Jun 16;2:41. doi: 10.3410/B2-41. F1000 Biol Rep. 2010. PMID: 20948792 Free PMC article.
-
The DYT6 Dystonia Protein THAP1 Regulates Myelination within the Oligodendrocyte Lineage.Dev Cell. 2017 Jul 10;42(1):52-67.e4. doi: 10.1016/j.devcel.2017.06.009. Dev Cell. 2017. PMID: 28697333 Free PMC article.
-
Genotype-phenotype correlations in THAP1 dystonia: molecular foundations and description of new cases.Parkinsonism Relat Disord. 2012 Jun;18(5):414-25. doi: 10.1016/j.parkreldis.2012.02.001. Epub 2012 Feb 28. Parkinsonism Relat Disord. 2012. PMID: 22377579 Free PMC article. Review.
-
Dimerization of the DYT6 dystonia protein, THAP1, requires residues within the coiled-coil domain.J Neurochem. 2011 Sep;118(6):1087-100. doi: 10.1111/j.1471-4159.2011.07386.x. Epub 2011 Aug 8. J Neurochem. 2011. PMID: 21752024 Free PMC article.
References
-
- Albanese A. Dystonia: clinical approach. Parkinsonism Relat Disord 2007;13(suppl 3):S356–S361. - PubMed
-
- Klein C, Ozelius LJ. Dystonia: clinical features, genetics, and treatment. Curr Opin Neurol 2002;15:491–497. - PubMed
-
- Ozelius LJ. Update on the genetics of primary torsion dystonia loci DYT6, DYT7, and DYT13 and the dystonia-plus locus DYT12. Adv Neurol 2004;94:109–112. - PubMed
-
- Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG. The pathophysiological basis of dystonias. Nat Rev Neurosci 2008;9:222–234. - PubMed
-
- Bressman SB, de Leon D, Raymond D, et al. The role of the DYT1 gene in secondary dystonia. Adv Neurol 1998;78:107–115. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases