

Adiponectin deficiency exacerbates cardiac dysfunction following pressure overload through disruption of an AMPK-dependent angiogenic response
- PMID: 20206634
- PMCID: PMC2885542
- DOI: 10.1016/j.yjmcc.2010.02.021
Adiponectin deficiency exacerbates cardiac dysfunction following pressure overload through disruption of an AMPK-dependent angiogenic response
Abstract
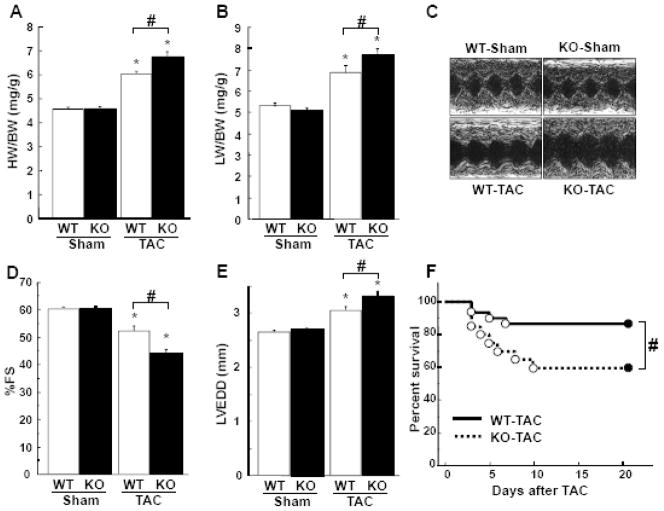
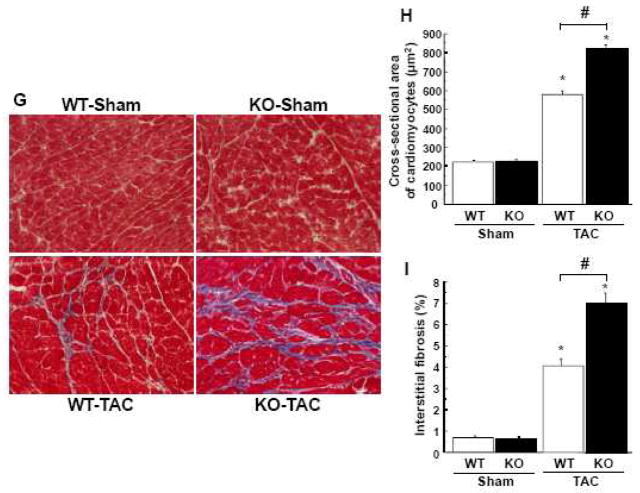
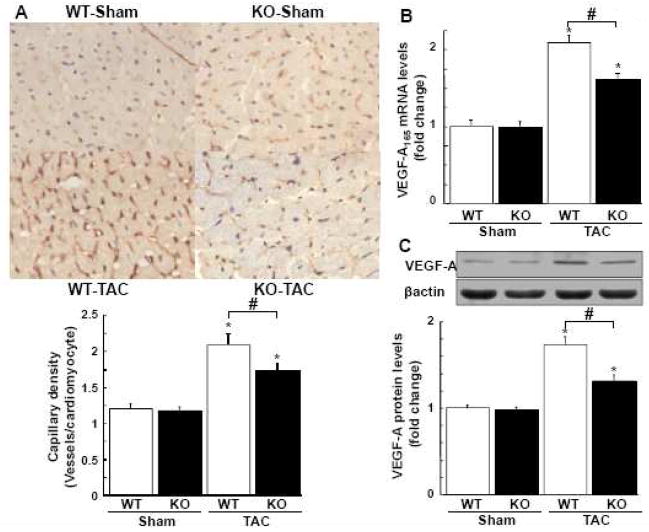
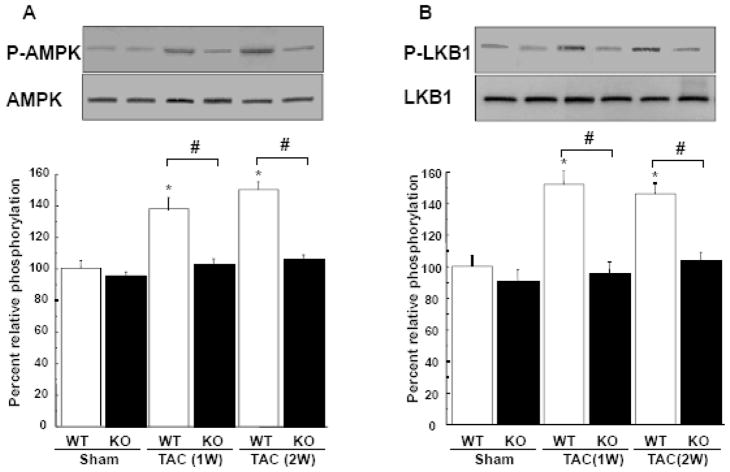
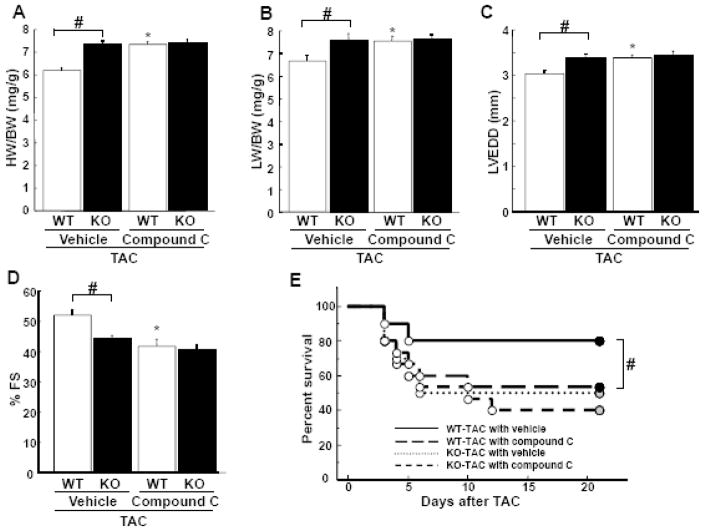
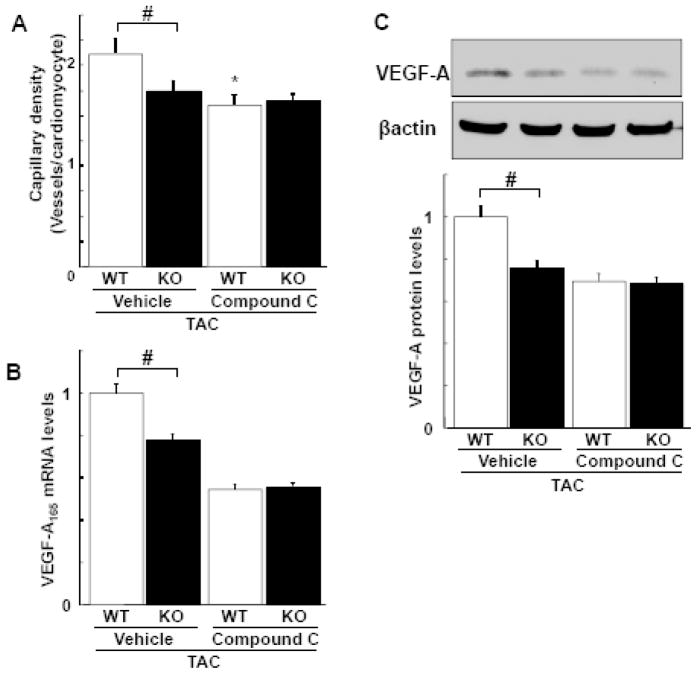
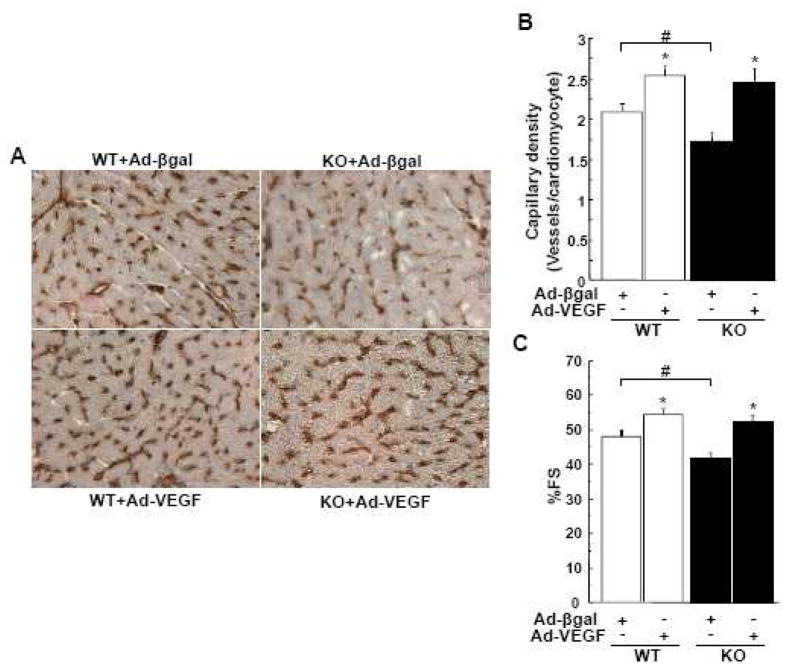
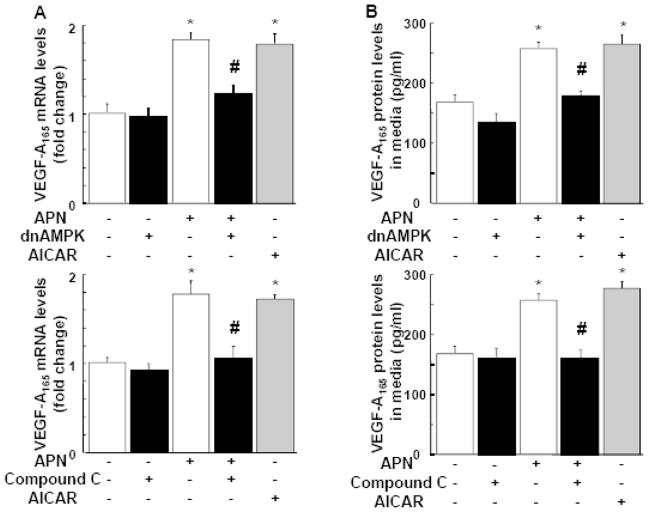
Although increasing evidence indicates that an adipokine adiponectin exerts protective actions on heart, its effects on coronary angiogenesis following pressure overload have not been examined previously. Because disruption of angiogenesis during heart growth leads to contractile dysfunction and heart failure, we hypothesized that adiponectin modulates cardiac remodeling in response to pressure overload through its ability to regulate adaptive angiogenesis. Adiponectin-knockout (APN-KO) and wild-type (WT) mice were subjected to pressure overload caused by transverse aortic constriction (TAC). APN-KO mice exhibited greater cardiac hypertrophy, pulmonary congestion, left ventricular (LV) interstitial fibrosis and LV systolic dysfunction after TAC surgery compared with WT mice. APN-KO mice also displayed reduced capillary density in the myocardium after TAC, which was accompanied by a significant decrease in expression of vascular endothelial growth factor (VEGF) and phosphorylation of AMP-activated protein kinase (AMPK). Inhibition of AMPK in WT mice resulted in aggravated LV systolic function, attenuated myocardial capillary density and decreased VEGF expression in response to TAC. The adverse effects of AMPK inhibition on cardiac function and angiogenic response following TAC were diminished in APN-KO mice relative to WT mice. Moreover, adenovirus-mediated VEGF delivery reversed the TAC-induced deficiencies in cardiac microvessel formation and ventricular function observed in the APN-KO mice. In cultured cardiac myocytes, adiponectin treatment stimulated VEGF production, which was inhibited by inactivation of AMPK signaling pathway. Collectively, these data show that adiponectin deficiency can accelerate the transition from cardiac hypertrophy to heart failure during pressure overload through disruption of AMPK-dependent angiogenic regulatory axis.
Figures
Similar articles
-
AMP activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice.Hypertension. 2008 Nov;52(5):918-24. doi: 10.1161/HYPERTENSIONAHA.108.114702. Epub 2008 Oct 6. Hypertension. 2008. PMID: 18838626 Free PMC article.
-
Adiponectin mediates cardioprotection in oxidative stress-induced cardiac myocyte remodeling.Am J Physiol Heart Circ Physiol. 2011 Sep;301(3):H984-93. doi: 10.1152/ajpheart.00428.2011. Epub 2011 Jun 10. Am J Physiol Heart Circ Physiol. 2011. PMID: 21666115 Free PMC article.
-
Connective tissue growth factor inhibition attenuates left ventricular remodeling and dysfunction in pressure overload-induced heart failure.Hypertension. 2014 Jun;63(6):1235-40. doi: 10.1161/HYPERTENSIONAHA.114.03279. Epub 2014 Mar 31. Hypertension. 2014. PMID: 24688123
-
AMPK in Cardiovascular Diseases.Exp Suppl. 2016;107:179-201. doi: 10.1007/978-3-319-43589-3_8. Exp Suppl. 2016. PMID: 27812981 Review.
-
[Effect of AMP-activated protein kinase on cardiovascular protection of adiponectin].Sheng Li Xue Bao. 2007 Oct 25;59(5):614-8. Sheng Li Xue Bao. 2007. PMID: 17940701 Review. Chinese.
Cited by
-
Adiponectin: mechanisms and new therapeutic approaches for restoring diabetic heart sensitivity to ischemic post-conditioning.Front Med. 2013 Sep;7(3):301-5. doi: 10.1007/s11684-013-0283-1. Epub 2013 Jul 20. Front Med. 2013. PMID: 23904036 Review.
-
Adiponectin enhances bone marrow mesenchymal stem cell resistance to flow shear stress through AMP-activated protein kinase signaling.Sci Rep. 2016 Jul 15;6:28752. doi: 10.1038/srep28752. Sci Rep. 2016. PMID: 27418435 Free PMC article.
-
Age-related cardiovascular disease and the beneficial effects of calorie restriction.Heart Fail Rev. 2012 Sep;17(4-5):707-19. doi: 10.1007/s10741-011-9293-8. Heart Fail Rev. 2012. PMID: 22095297 Review.
-
Tachycardia pacing induces myocardial neovascularization and mobilizes circulating endothelial progenitor cells partly via SDF-1 pathway in canines.Heart Vessels. 2016 Feb;31(2):230-40. doi: 10.1007/s00380-014-0613-5. Epub 2014 Dec 10. Heart Vessels. 2016. PMID: 25491934
-
Adiponectin downregulation is associated with volume overload-induced myocyte dysfunction in rats.Acta Pharmacol Sin. 2016 Feb;37(2):187-95. doi: 10.1038/aps.2015.84. Epub 2015 Nov 30. Acta Pharmacol Sin. 2016. PMID: 26616727 Free PMC article.
References
-
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiography determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–6. - PubMed
-
- Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–9. - PubMed
-
- Luedde M, Katus HA, Frey N. Novel molecular targets in the treatment of cardiac hypertrophy. Recent Pat Cardiovasc Drug Discov. 2006;1:1–20. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HL77774/HL/NHLBI NIH HHS/United States
- R01 HL086785-18/HL/NHLBI NIH HHS/United States
- R01 HL086785/HL/NHLBI NIH HHS/United States
- P01 HL081587-05/HL/NHLBI NIH HHS/United States
- R01 AG015052/AG/NIA NIH HHS/United States
- AG15052/AG/NIA NIH HHS/United States
- R37 AG015052-12/AG/NIA NIH HHS/United States
- R37 AG015052/AG/NIA NIH HHS/United States
- HL81587/HL/NHLBI NIH HHS/United States
- R01 HL077774/HL/NHLBI NIH HHS/United States
- P01 HL081587/HL/NHLBI NIH HHS/United States
- R01 HL077774-05/HL/NHLBI NIH HHS/United States
- HL86785/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
