

Anticancer Role of PPARgamma Agonists in Hematological Malignancies Found in the Vasculature, Marrow, and Eyes
- PMID: 20204067
- PMCID: PMC2829627
- DOI: 10.1155/2010/814609
Anticancer Role of PPARgamma Agonists in Hematological Malignancies Found in the Vasculature, Marrow, and Eyes
Abstract
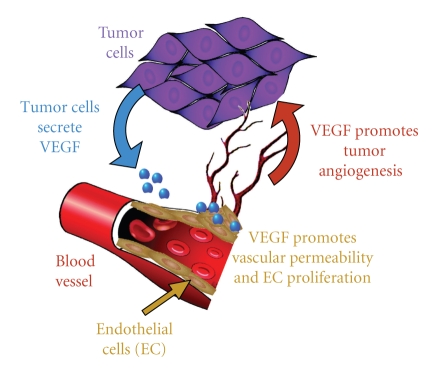
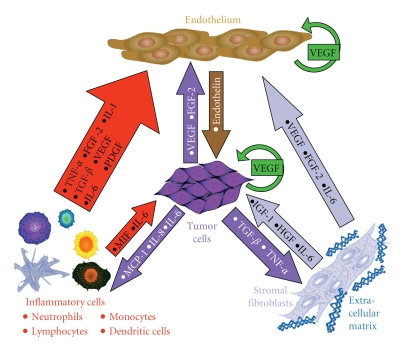
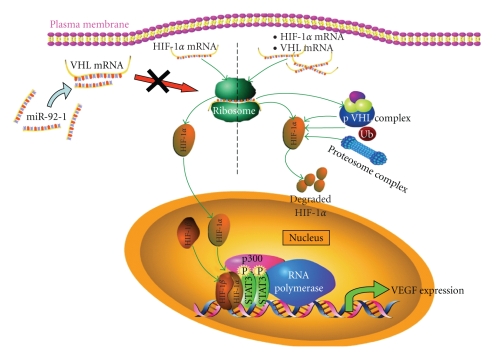
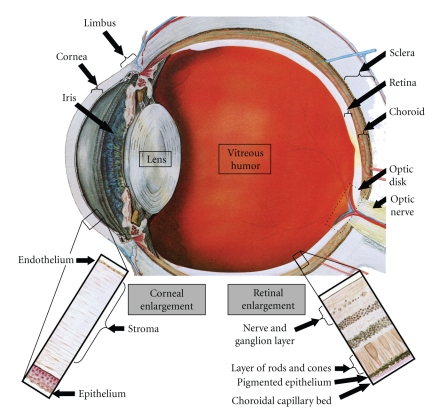
The use of targeted cancer therapies in combination with conventional chemotherapeutic agents and/or radiation treatment has increased overall survival of cancer patients. However, longer survival is accompanied by increased incidence of comorbidities due, in part, to drug side effects and toxicities. It is well accepted that inflammation and tumorigenesis are linked. Because peroxisome proliferator-activated receptor (PPAR)-gamma agonists are potent mediators of anti-inflammatory responses, it was a logical extension to examine the role of PPARgamma agonists in the treatment and prevention of cancer. This paper has two objectives: first to highlight the potential uses for PPARgamma agonists in anticancer therapy with special emphasis on their role when used as adjuvant or combined therapy in the treatment of hematological malignancies found in the vasculature, marrow, and eyes, and second, to review the potential role PPARgamma and/or its ligands may have in modulating cancer-associated angiogenesis and tumor-stromal microenvironment crosstalk in bone marrow.
Figures


Similar articles
-
Evidence that peroxisome proliferator-activated receptor γ suppresses squamous carcinogenesis through anti-inflammatory signaling and regulation of the immune response.Mol Carcinog. 2019 Sep;58(9):1589-1601. doi: 10.1002/mc.23041. Epub 2019 May 20. Mol Carcinog. 2019. PMID: 31111568 Review.
-
PPARγ Agonists in Combination Cancer Therapies.Curr Cancer Drug Targets. 2020;20(3):197-215. doi: 10.2174/1568009619666191209102015. Curr Cancer Drug Targets. 2020. PMID: 31814555 Review.
-
Combined treatment with peroxisome proliferator-activated receptor (PPAR) gamma ligands and gamma radiation induces apoptosis by PPARγ-independent up-regulation of reactive oxygen species-induced deoxyribonucleic acid damage signals in non-small cell lung cancer cells.Int J Radiat Oncol Biol Phys. 2013 Apr 1;85(5):e239-48. doi: 10.1016/j.ijrobp.2012.11.040. Epub 2013 Jan 17. Int J Radiat Oncol Biol Phys. 2013. PMID: 23332223
-
Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) as a Target for Concurrent Management of Diabetes and Obesity-Related Cancer.Curr Pharm Des. 2017;23(25):3677-3688. doi: 10.2174/1381612823666170704125104. Curr Pharm Des. 2017. PMID: 28677503 Review.
-
PPARgamma as a therapeutic target for tumor angiogenesis and metastasis.Cancer Biol Ther. 2005 Jul;4(7):687-93. doi: 10.4161/cbt.4.7.2014. Epub 2005 Jul 13. Cancer Biol Ther. 2005. PMID: 16082179 Review.
Cited by
-
Telmisartan counteracts TGF-β1 induced epithelial-to-mesenchymal transition via PPAR-γ in human proximal tubule epithelial cells.Int J Clin Exp Pathol. 2012;5(6):522-9. Epub 2012 Jul 17. Int J Clin Exp Pathol. 2012. PMID: 22949934 Free PMC article.
-
The complexities of obesity and diabetes with the development and progression of pancreatic cancer.Biochim Biophys Acta. 2011 Apr;1815(2):135-46. doi: 10.1016/j.bbcan.2010.11.003. Epub 2010 Dec 1. Biochim Biophys Acta. 2011. PMID: 21129444 Free PMC article. Review.
-
Methyl 2-cyano-3,11-dioxo-18-olean-1,12-dien-30-oate (CDODA-Me), a derivative of glycyrrhetinic acid, functions as a potent angiogenesis inhibitor.J Pharmacol Exp Ther. 2010 Oct;335(1):172-9. doi: 10.1124/jpet.110.171066. Epub 2010 Jul 14. J Pharmacol Exp Ther. 2010. PMID: 20631299 Free PMC article.
-
Antifibrotic Actions of Peroxisome Proliferator-Activated Receptor γ Ligands in Corneal Fibroblasts Are Mediated by β-Catenin-Regulated Pathways.Am J Pathol. 2017 Aug;187(8):1660-1669. doi: 10.1016/j.ajpath.2017.04.002. Epub 2017 Jun 10. Am J Pathol. 2017. PMID: 28606794 Free PMC article.
-
Inhibitory effects of PPARγ ligands on TGF-β1-induced corneal myofibroblast transformation.Am J Pathol. 2014 May;184(5):1429-45. doi: 10.1016/j.ajpath.2014.01.026. Epub 2014 Mar 17. Am J Pathol. 2014. PMID: 24650561 Free PMC article.
References
-
- Lazar MA. PPARγ, 10 years later. Biochimie. 2005;87(1):9–13. - PubMed
-
- Sharma AM, Staels B. Review: peroxisome proliferator-activated receptor γ and adipose tissue—understanding obesity-related changes in regulation of lipid and glucose metabolism. Journal of Clinical Endocrinology and Metabolism. 2007;92(2):386–395. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
