

Cdk5 nuclear localization is p27-dependent in nerve cells: implications for cell cycle suppression and caspase-3 activation
- PMID: 20189989
- PMCID: PMC2859566
- DOI: 10.1074/jbc.M109.068262
Cdk5 nuclear localization is p27-dependent in nerve cells: implications for cell cycle suppression and caspase-3 activation
Abstract
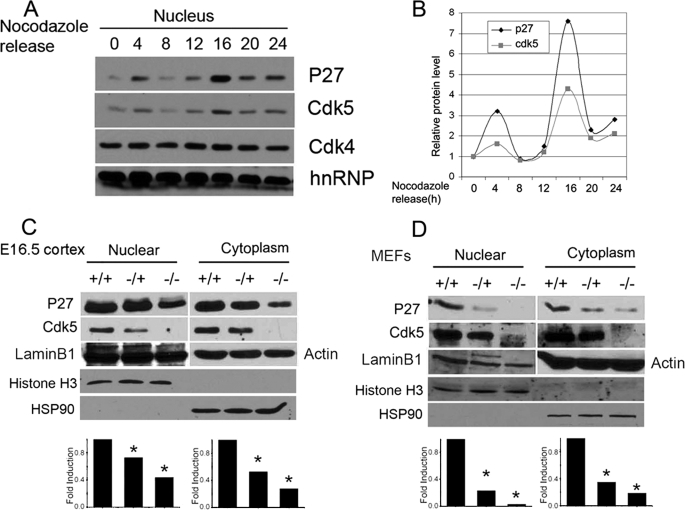
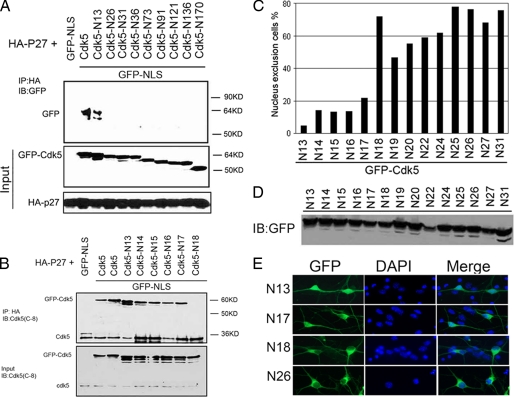
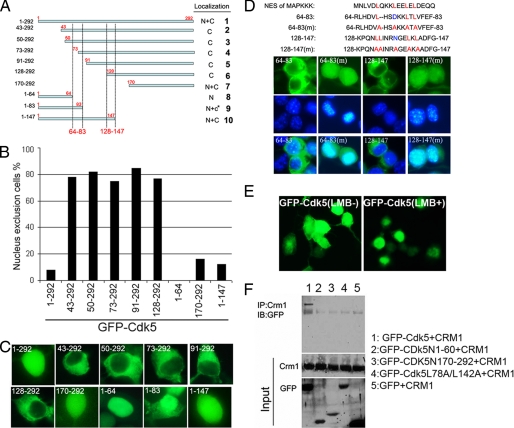
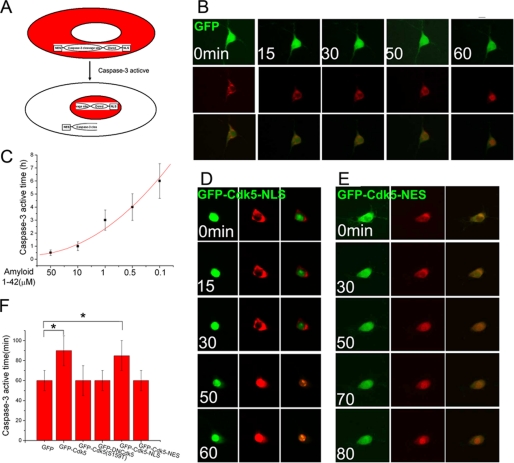
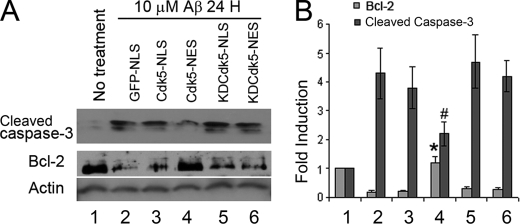
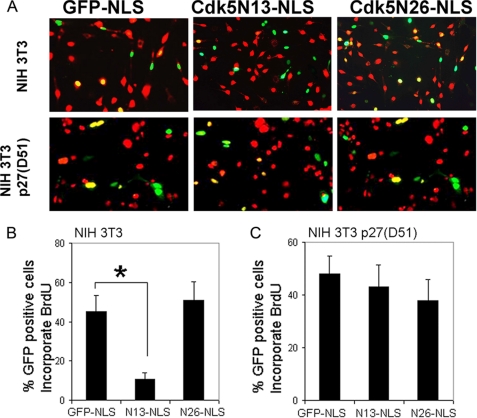
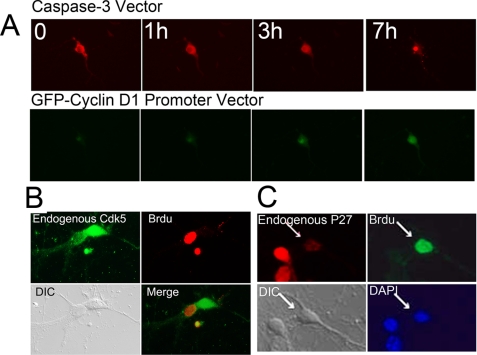
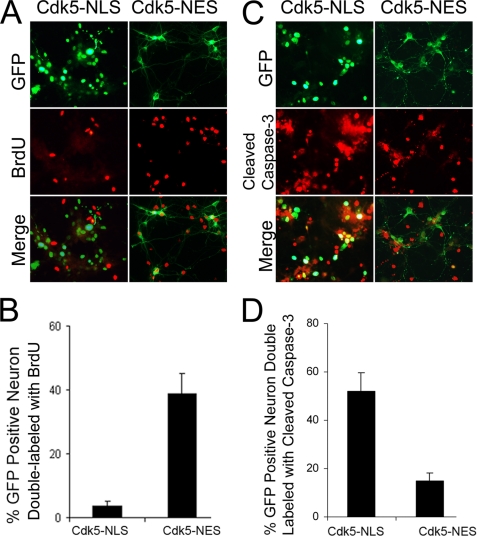
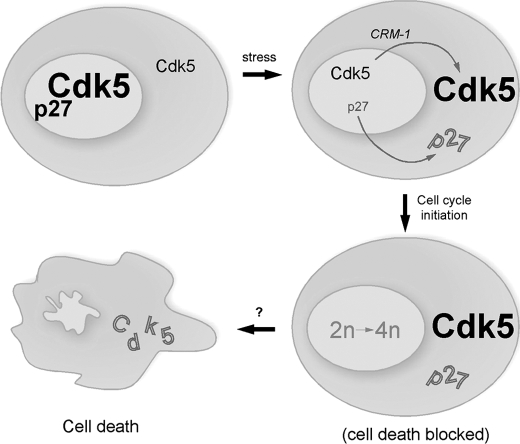
Initiation of a cell cycle in an adult neuron leads to cell death, placing great importance on the mechanisms that normally suppress the neuronal cell cycle. We have previously shown that the cyclin-dependent kinase Cdk5 is an important part of this process, but only when it is present in the nucleus. We report here that Cdk5 nuclear localization relies on its binding to the cyclin-dependent kinase inhibitor p27. Cdk5 has no intrinsic nuclear localization signal; in the absence of p27, two weak nuclear export signals that bind CRM1 cause it to shuttle to the cytoplasm. When a neuron is subjected to stress, such as exposure to beta-amyloid, the Cdk5-p27 interaction is lost, reducing Cdk5 levels in the nucleus and depriving the neuron of a major cell cycle suppression mechanism. Caspase-3 is activated within hours, but death is not immediate; elevated levels of cytoplasmic Cdk5 appear to retard neuronal death by a mechanism that may involve Bcl2. These data suggest a model in which Cdk5 exerts a double protective function in neurons: chronically suppressing the cell cycle when located in the nucleus and transiently delaying cell death in the cytoplasm.
Figures
Similar articles
-
Nucleocytoplasmic Cdk5 is involved in neuronal cell cycle and death in post-mitotic neurons.Cell Cycle. 2011 Apr 15;10(8):1208-14. doi: 10.4161/cc.10.8.15328. Epub 2011 Apr 15. Cell Cycle. 2011. PMID: 21415596
-
Cdk5 and its substrates, Dcx and p27kip1, regulate cytoplasmic dilation formation and nuclear elongation in migrating neurons.Development. 2014 Sep;141(18):3540-50. doi: 10.1242/dev.111294. Development. 2014. PMID: 25183872
-
Suppression of nucleocytoplasmic p27Kip1 export attenuates CDK4-mediated neuronal death induced by status epilepticus.Neurosci Res. 2018 Jul;132:46-52. doi: 10.1016/j.neures.2017.10.001. Epub 2017 Oct 10. Neurosci Res. 2018. PMID: 29024678
-
Regulation and role of cyclin-dependent kinase activity in neuronal survival and death.J Neurochem. 2010 Dec;115(6):1309-21. doi: 10.1111/j.1471-4159.2010.07050.x. Epub 2010 Nov 4. J Neurochem. 2010. PMID: 21044075 Review.
-
CDK5: an oncogene or an anti-oncogene: location location location.Mol Cancer. 2023 Nov 23;22(1):186. doi: 10.1186/s12943-023-01895-8. Mol Cancer. 2023. PMID: 37993880 Free PMC article. Review.
Cited by
-
The roles of Cdk5-mediated subcellular localization of FOXO1 in neuronal death.J Neurosci. 2015 Feb 11;35(6):2624-35. doi: 10.1523/JNEUROSCI.3051-14.2015. J Neurosci. 2015. PMID: 25673854 Free PMC article.
-
Regulation of sirtuin function by posttranslational modifications.Front Pharmacol. 2012 Feb 28;3:29. doi: 10.3389/fphar.2012.00029. eCollection 2012. Front Pharmacol. 2012. PMID: 22403547 Free PMC article.
-
Cdk5 regulates IP3R1-mediated Ca2+ dynamics and Ca2+-mediated cell proliferation.Cell Mol Life Sci. 2022 Aug 24;79(9):495. doi: 10.1007/s00018-022-04515-8. Cell Mol Life Sci. 2022. PMID: 36001172 Free PMC article.
-
Cdk5 controls IL-2 gene expression via repression of the mSin3a-HDAC complex.Cell Cycle. 2015;14(8):1327-36. doi: 10.4161/15384101.2014.987621. Cell Cycle. 2015. PMID: 25785643 Free PMC article.
-
Role of a cdk5-associated protein, p35, in herpes simplex virus type 1 replication in vivo.J Neurovirol. 2010 Oct;16(5):405-9. doi: 10.3109/13550284.2010.513030. J Neurovirol. 2010. PMID: 20839922 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
