

Absolute binding free energy calculations: on the accuracy of computational scoring of protein-ligand interactions
- PMID: 20186976
- PMCID: PMC2868600
- DOI: 10.1002/prot.22687
Absolute binding free energy calculations: on the accuracy of computational scoring of protein-ligand interactions
Abstract
Calculating the absolute binding free energies is a challenging task. Reliable estimates of binding free energies should provide a guide for rational drug design. It should also provide us with deeper understanding of the correlation between protein structure and its function. Further applications may include identifying novel molecular scaffolds and optimizing lead compounds in computer-aided drug design. Available options to evaluate the absolute binding free energies range from the rigorous but expensive free energy perturbation to the microscopic linear response approximation (LRA/beta version) and related approaches including the linear interaction energy (LIE) to the more approximated and considerably faster scaled protein dipoles Langevin dipoles (PDLD/S-LRA version) as well as the less rigorous molecular mechanics Poisson-Boltzmann/surface area (MM/PBSA) and generalized born/surface area (MM/GBSA) to the less accurate scoring functions. There is a need for an assessment of the performance of different approaches in terms of computer time and reliability. We present a comparative study of the LRA/beta, the LIE, the PDLD/S-LRA/beta, and the more widely used MM/PBSA and assess their abilities to estimate the absolute binding energies. The LRA and LIE methods perform reasonably well but require specialized parameterization for the nonelectrostatic term. The PDLD/S-LRA/beta performs effectively without the need of reparameterization. Our assessment of the MM/PBSA is less optimistic. This approach appears to provide erroneous estimates of the absolute binding energies because of its incorrect entropies and the problematic treatment of electrostatic energies. Overall, the PDLD/S-LRA/beta appears to offer an appealing option for the final stages of massive screening approaches.
Proteins 2010. (c) 2010 Wiley-Liss, Inc.
Figures




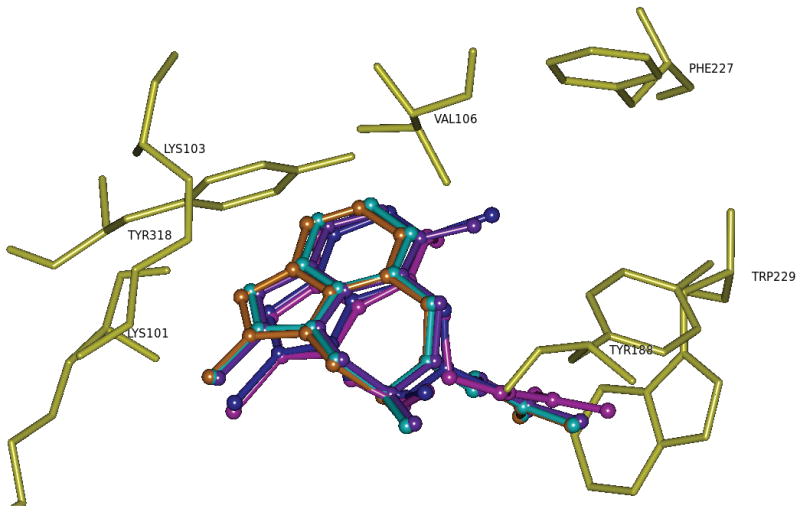
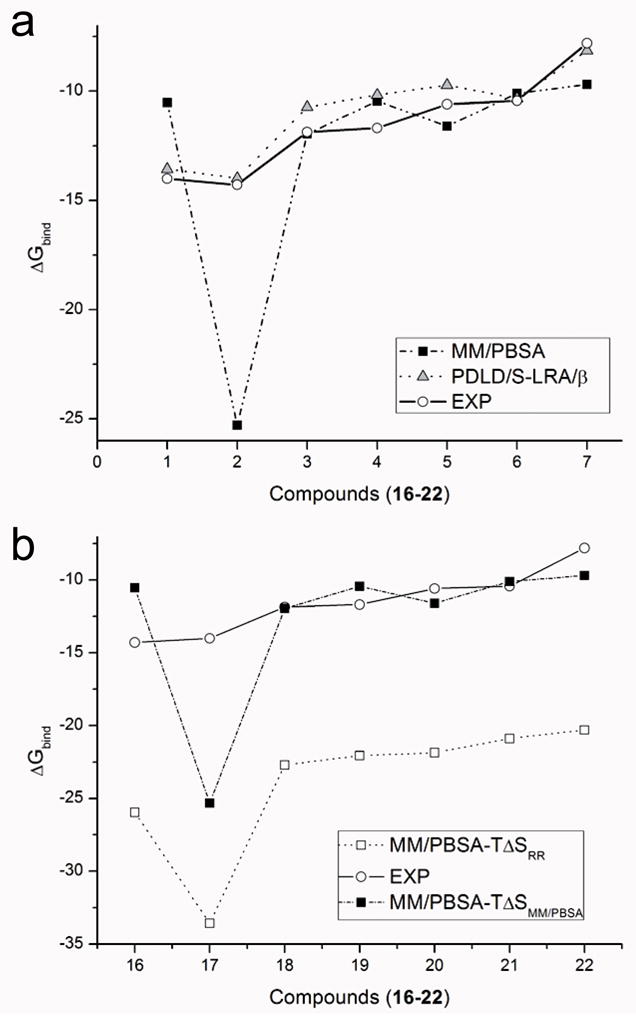
Similar articles
-
Examining methods for calculations of binding free energies: LRA, LIE, PDLD-LRA, and PDLD/S-LRA calculations of ligands binding to an HIV protease.Proteins. 2000 Jun 1;39(4):393-407. Proteins. 2000. PMID: 10813821
-
Comparison of end-point continuum-solvation methods for the calculation of protein-ligand binding free energies.Proteins. 2012 May;80(5):1326-42. doi: 10.1002/prot.24029. Epub 2012 Feb 13. Proteins. 2012. PMID: 22274991
-
A head-to-head comparison of MM/PBSA and MM/GBSA in predicting binding affinities for the CB1 cannabinoid ligands.J Mol Model. 2024 Oct 31;30(11):390. doi: 10.1007/s00894-024-06189-4. J Mol Model. 2024. PMID: 39480515
-
Free energy calculations to estimate ligand-binding affinities in structure-based drug design.Curr Pharm Des. 2014;20(20):3323-37. doi: 10.2174/13816128113199990604. Curr Pharm Des. 2014. PMID: 23947646 Review.
-
The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities.Expert Opin Drug Discov. 2015 May;10(5):449-61. doi: 10.1517/17460441.2015.1032936. Epub 2015 Apr 2. Expert Opin Drug Discov. 2015. PMID: 25835573 Free PMC article. Review.
Cited by
-
Molecular Mechanisms of DNA Replication and Repair Machinery: Insights from Microscopic Simulations.Adv Theory Simul. 2019 May;2(5):1800191. doi: 10.1002/adts.201800191. Epub 2019 Feb 12. Adv Theory Simul. 2019. PMID: 31728433 Free PMC article.
-
The Nature of Functional Features of Different Classes of G-Protein-Coupled Receptors.Biology (Basel). 2022 Dec 16;11(12):1839. doi: 10.3390/biology11121839. Biology (Basel). 2022. PMID: 36552350 Free PMC article.
-
Novel MscL agonists that allow multiple antibiotics cytoplasmic access activate the channel through a common binding site.PLoS One. 2020 Jan 24;15(1):e0228153. doi: 10.1371/journal.pone.0228153. eCollection 2020. PLoS One. 2020. PMID: 31978161 Free PMC article.
-
Binding affinities in the SAMPL3 trypsin and host-guest blind tests estimated with the MM/PBSA and LIE methods.J Comput Aided Mol Des. 2012 May;26(5):527-41. doi: 10.1007/s10822-011-9524-z. Epub 2011 Dec 25. J Comput Aided Mol Des. 2012. PMID: 22198518
-
Targeting HCV polymerase: a structural and dynamic perspective into the mechanism of selective covalent inhibition.RSC Adv. 2018 Dec 18;8(73):42210-42222. doi: 10.1039/c8ra07346e. eCollection 2018 Dec 12. RSC Adv. 2018. PMID: 35558797 Free PMC article.
References
-
- Warshel A. Electrostatic basis of structure-function correlation in proteins. Acc Chem Res. 1981;14:284–290.
-
- Warshel A, Åqvist J. Electrostatic Energy and Macromolecular Function. Ann Rev Biophys Chem. 1991;20:267–298. - PubMed
-
- Huang N, MPJ Physics-based methods for studying protein-ligand interactions. Curr Opin Drug Discov Devel. 2007;10:325–331. - PubMed
-
- Raha K, Merz KM., Jr Calculating binding free energy in protein-ligand interaction. Annual Reports in Computational Chemistry. 2005;1:113–130.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
