

Activation of p38 MAPK pathway in the skull abnormalities of Apert syndrome Fgfr2(+P253R) mice
- PMID: 20175913
- PMCID: PMC2838826
- DOI: 10.1186/1471-213X-10-22
Activation of p38 MAPK pathway in the skull abnormalities of Apert syndrome Fgfr2(+P253R) mice
Abstract
Background: Apert syndrome is characterized by craniosynostosis and limb abnormalities and is primarily caused by FGFR2 +/P253R and +/S252W mutations. The former mutation is present in approximately one third whereas the latter mutation is present in two-thirds of the patients with this condition. We previously reported an inbred transgenic mouse model with the Fgfr2 +/S252W mutation on the C57BL/6J background for Apert syndrome. Here we present a mouse model for the Fgfr2+/P253R mutation.
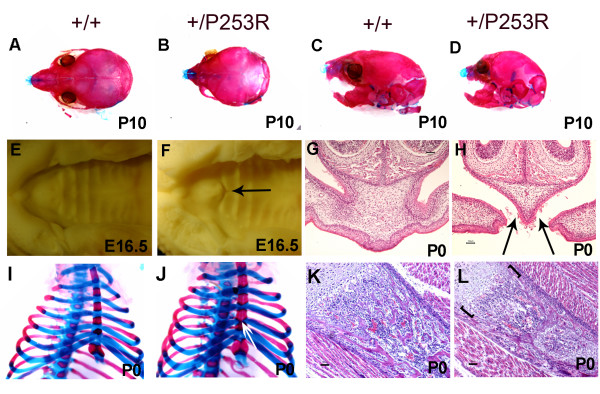
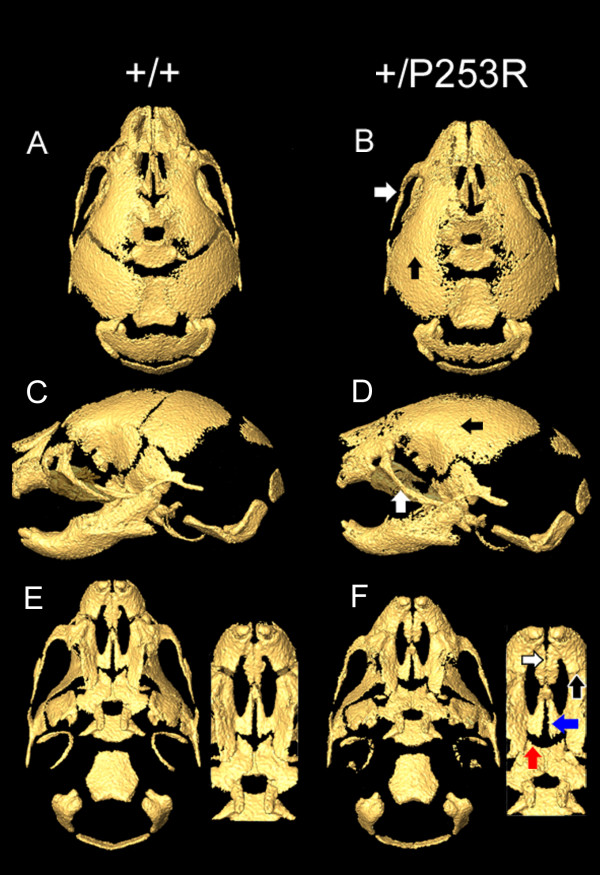
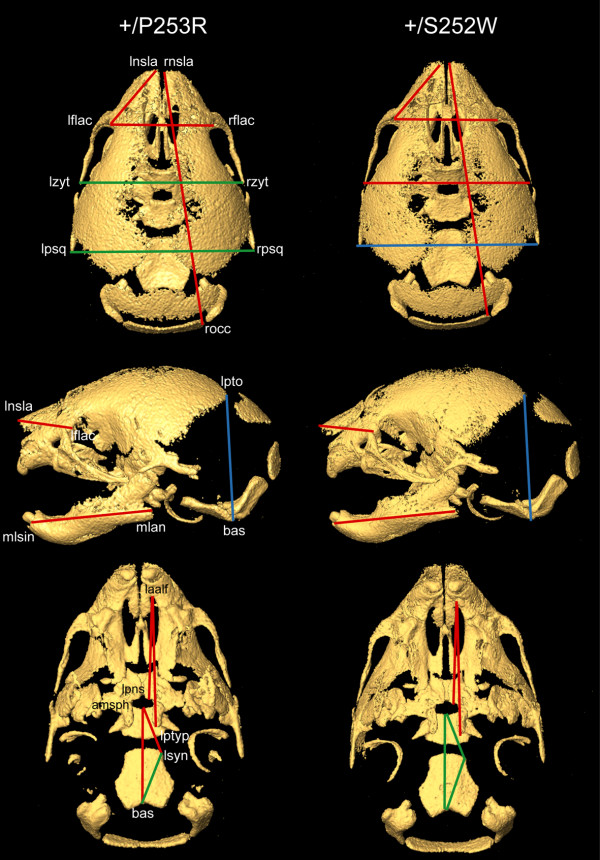
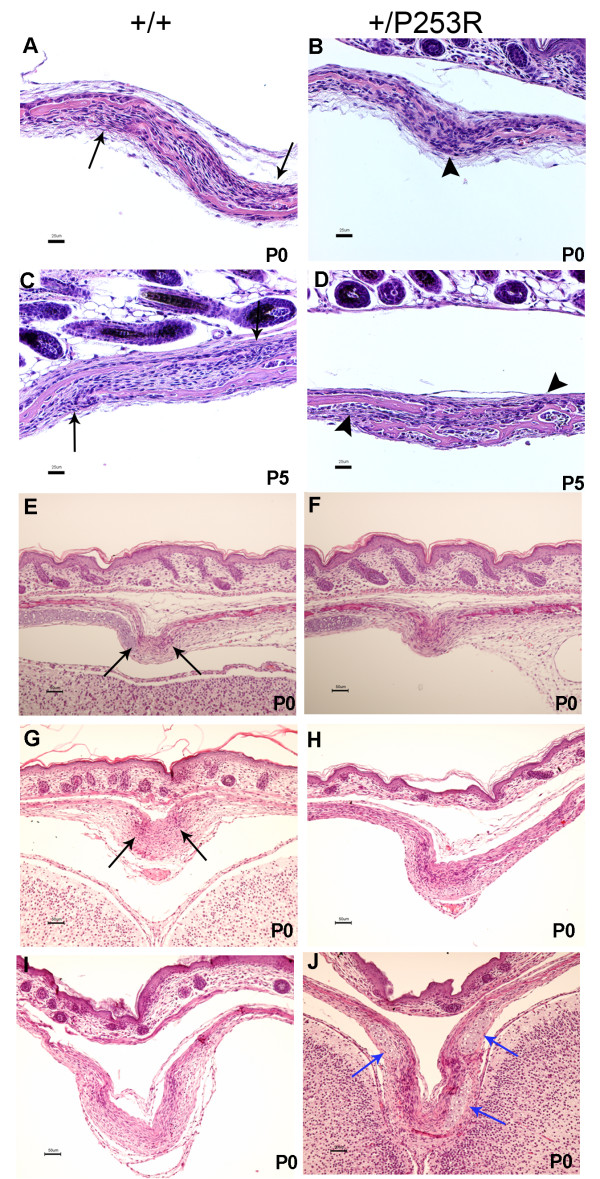
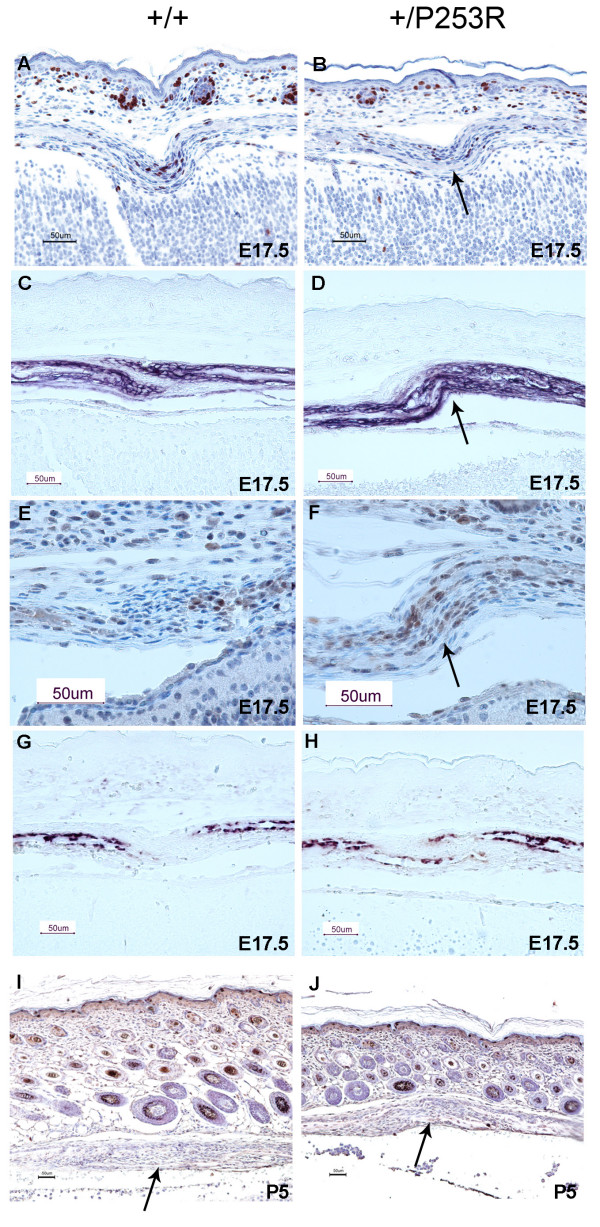
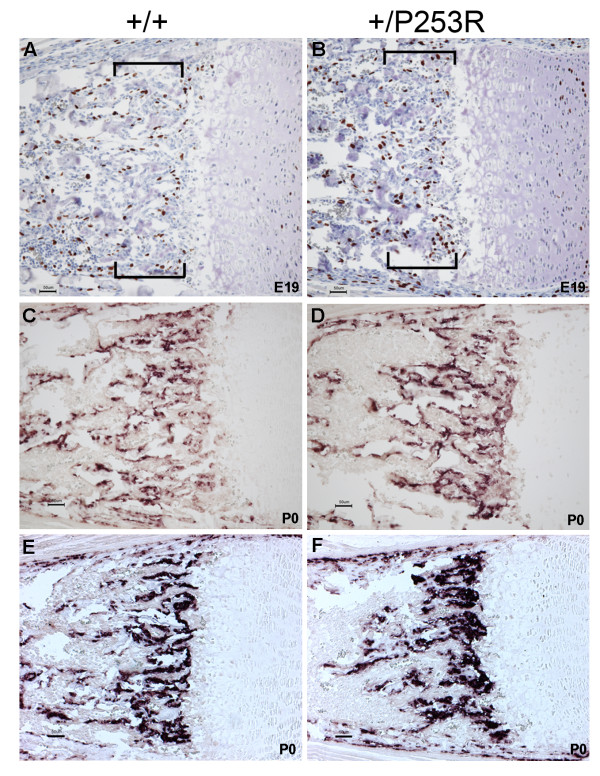
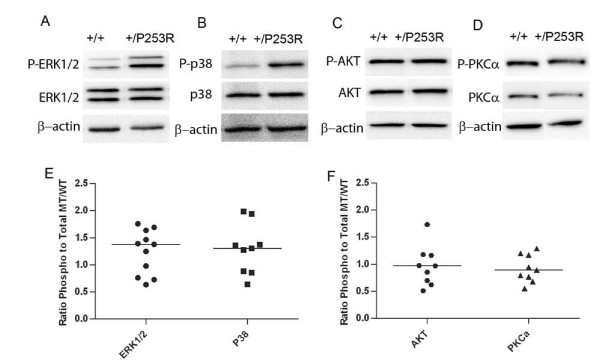
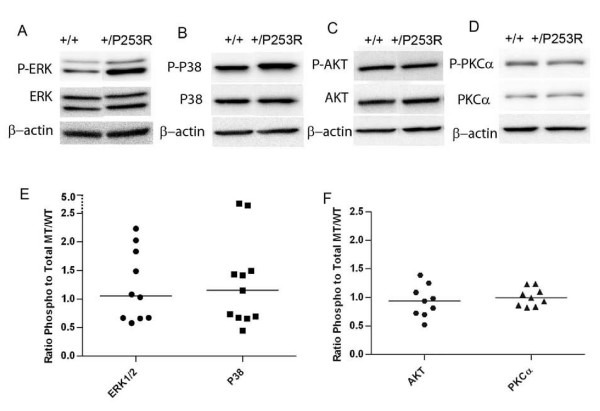
Results: We generated inbred Fgfr2(+/P253R) mice on the same C56BL/6J genetic background and analyzed their skeletal abnormalities. 3D micro-CT scans of the skulls of the Fgfr2(+/P253R) mice revealed that the skull length was shortened with the length of the anterior cranial base significantly shorter than that of the Fgfr2(+/S252W) mice at P0. The Fgfr2(+/P253R) mice presented with synostosis of the coronal suture and proximate fronts with disorganized cellularity in sagittal and lambdoid sutures. Abnormal osteogenesis and proliferation were observed at the developing coronal suture and long bones of the Fgfr2(+/P253R) mice as in the Fgfr2(+/S252W) mice. Activation of mitogen-activated protein kinases (MAPK) was observed in the Fgfr2(+/P253R) neurocranium with an increase in phosphorylated p38 as well as ERK1/2, whereas phosphorylated AKT and PKCalpha were not obviously changed as compared to those of wild-type controls. There were localized phenotypic and molecular variations among individual embryos with different mutations and among those with the same mutation.
Conclusions: Our in vivo studies demonstrated that the Fgfr2 +/P253R mutation resulted in mice with cranial features that resemble those of the Fgfr2(+/S252W) mice and human Apert syndrome. Activated p38 in addition to the ERK1/2 signaling pathways may mediate the mutant neurocranial phenotype. Though Apert syndrome is traditionally thought to be a consistent phenotype, our results suggest localized and regional variations in the phenotypes that characterize Apert syndrome.
Figures
Similar articles
-
A Pro253Arg mutation in fibroblast growth factor receptor 2 (Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis.Bone. 2008 Apr;42(4):631-43. doi: 10.1016/j.bone.2007.11.019. Epub 2008 Jan 31. Bone. 2008. PMID: 18242159
-
Deformed Skull Morphology Is Caused by the Combined Effects of the Maldevelopment of Calvarias, Cranial Base and Brain in FGFR2-P253R Mice Mimicking Human Apert Syndrome.Int J Biol Sci. 2017 Jan 1;13(1):32-45. doi: 10.7150/ijbs.16287. eCollection 2017. Int J Biol Sci. 2017. PMID: 28123344 Free PMC article.
-
Beyond the closed suture in apert syndrome mouse models: evidence of primary effects of FGFR2 signaling on facial shape at birth.Dev Dyn. 2010 Nov;239(11):3058-71. doi: 10.1002/dvdy.22414. Dev Dyn. 2010. PMID: 20842696 Free PMC article.
-
Cleft Palate in Apert Syndrome.J Dev Biol. 2022 Aug 11;10(3):33. doi: 10.3390/jdb10030033. J Dev Biol. 2022. PMID: 35997397 Free PMC article. Review.
-
Insights and future directions of potential genetic therapy for Apert syndrome: A systematic review.Gene Ther. 2021 Nov;28(10-11):620-633. doi: 10.1038/s41434-021-00238-w. Epub 2021 Feb 22. Gene Ther. 2021. PMID: 33619359 Review.
Cited by
-
Quantification of shape and cell polarity reveals a novel mechanism underlying malformations resulting from related FGF mutations during facial morphogenesis.Hum Mol Genet. 2013 Dec 20;22(25):5160-72. doi: 10.1093/hmg/ddt369. Epub 2013 Aug 1. Hum Mol Genet. 2013. PMID: 23906837 Free PMC article.
-
Research advances in Apert syndrome.J Oral Biol Craniofac Res. 2018 Sep-Dec;8(3):194-199. doi: 10.1016/j.jobcr.2017.05.006. Epub 2017 May 25. J Oral Biol Craniofac Res. 2018. PMID: 30191107 Free PMC article. Review.
-
Limb reduction in an Esco2 cohesinopathy mouse model is mediated by p53-dependent apoptosis and vascular disruption.Nat Commun. 2024 Aug 21;15(1):7154. doi: 10.1038/s41467-024-51328-3. Nat Commun. 2024. PMID: 39168984 Free PMC article.
-
Hand in glove: brain and skull in development and dysmorphogenesis.Acta Neuropathol. 2013 Apr;125(4):469-89. doi: 10.1007/s00401-013-1104-y. Epub 2013 Mar 23. Acta Neuropathol. 2013. PMID: 23525521 Free PMC article. Review.
-
Mouse models of Apert syndrome.Childs Nerv Syst. 2012 Sep;28(9):1505-10. doi: 10.1007/s00381-012-1872-z. Epub 2012 Aug 8. Childs Nerv Syst. 2012. PMID: 22872267
References
-
- Olsen SK, Ibrahimi OA, Raucci A, Zhang F, Eliseenkova AV, Yayon A, Basilico C, Linhardt RJ, Schlessinger J, Mohammadi M. Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proc Natl Acad Sci USA. 2004;101:935–940. doi: 10.1073/pnas.0307287101. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
