

Fast and SNP-tolerant detection of complex variants and splicing in short reads
- PMID: 20147302
- PMCID: PMC2844994
- DOI: 10.1093/bioinformatics/btq057
Fast and SNP-tolerant detection of complex variants and splicing in short reads
Abstract
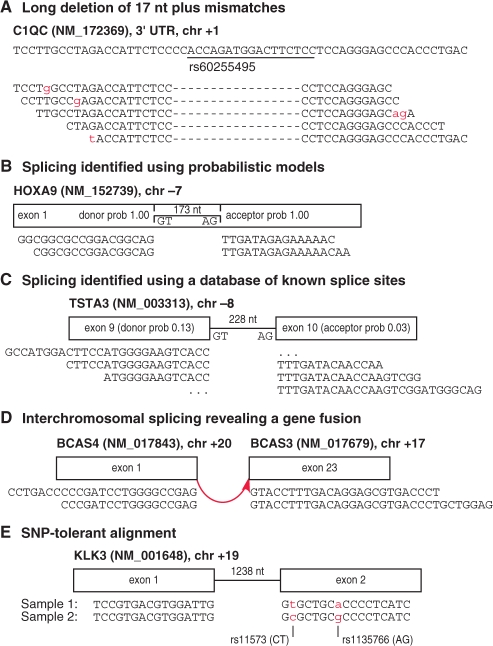
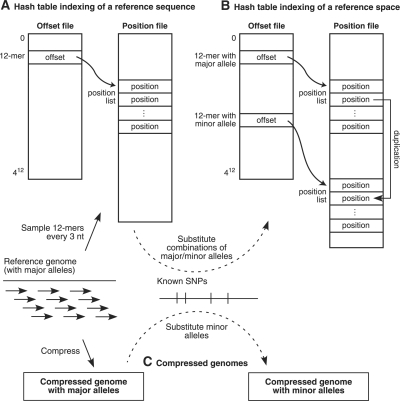
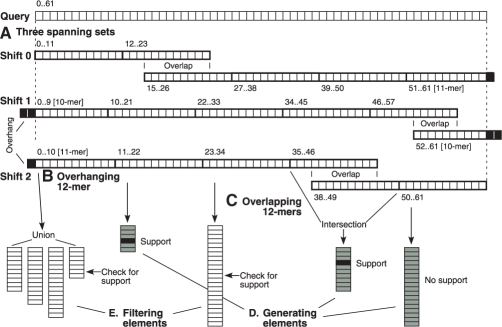
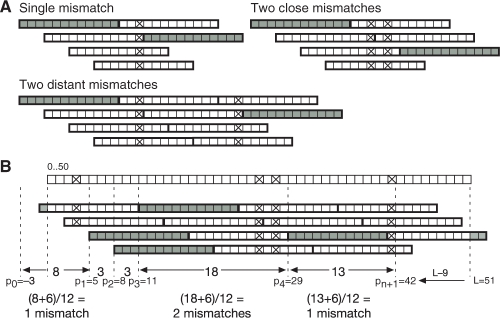
Motivation: Next-generation sequencing captures sequence differences in reads relative to a reference genome or transcriptome, including splicing events and complex variants involving multiple mismatches and long indels. We present computational methods for fast detection of complex variants and splicing in short reads, based on a successively constrained search process of merging and filtering position lists from a genomic index. Our methods are implemented in GSNAP (Genomic Short-read Nucleotide Alignment Program), which can align both single- and paired-end reads as short as 14 nt and of arbitrarily long length. It can detect short- and long-distance splicing, including interchromosomal splicing, in individual reads, using probabilistic models or a database of known splice sites. Our program also permits SNP-tolerant alignment to a reference space of all possible combinations of major and minor alleles, and can align reads from bisulfite-treated DNA for the study of methylation state.
Results: In comparison testing, GSNAP has speeds comparable to existing programs, especially in reads of > or=70 nt and is fastest in detecting complex variants with four or more mismatches or insertions of 1-9 nt and deletions of 1-30 nt. Although SNP tolerance does not increase alignment yield substantially, it affects alignment results in 7-8% of transcriptional reads, typically by revealing alternate genomic mappings for a read. Simulations of bisulfite-converted DNA show a decrease in identifying genomic positions uniquely in 6% of 36 nt reads and 3% of 70 nt reads.
Availability: Source code in C and utility programs in Perl are freely available for download as part of the GMAP package at http://share.gene.com/gmap.
Figures
Similar articles
-
GMAP and GSNAP for Genomic Sequence Alignment: Enhancements to Speed, Accuracy, and Functionality.Methods Mol Biol. 2016;1418:283-334. doi: 10.1007/978-1-4939-3578-9_15. Methods Mol Biol. 2016. PMID: 27008021
-
Fast and SNP-aware short read alignment with SALT.BMC Bioinformatics. 2021 Aug 25;22(Suppl 9):172. doi: 10.1186/s12859-021-04088-6. BMC Bioinformatics. 2021. PMID: 34433415 Free PMC article.
-
Fast and accurate short read alignment with Burrows-Wheeler transform.Bioinformatics. 2009 Jul 15;25(14):1754-60. doi: 10.1093/bioinformatics/btp324. Epub 2009 May 18. Bioinformatics. 2009. PMID: 19451168 Free PMC article.
-
Read-Split-Run: an improved bioinformatics pipeline for identification of genome-wide non-canonical spliced regions using RNA-Seq data.BMC Genomics. 2016 Aug 22;17 Suppl 7(Suppl 7):503. doi: 10.1186/s12864-016-2896-7. BMC Genomics. 2016. PMID: 27556805 Free PMC article.
-
Challenges in identifying large germline structural variants for clinical use by long read sequencing.Comput Struct Biotechnol J. 2019 Dec 23;18:83-92. doi: 10.1016/j.csbj.2019.11.008. eCollection 2020. Comput Struct Biotechnol J. 2019. PMID: 32099591 Free PMC article. Review.
Cited by
-
A thesaurus of genetic variation for interrogation of repetitive genomic regions.Nucleic Acids Res. 2015 May 26;43(10):e68. doi: 10.1093/nar/gkv178. Epub 2015 Mar 27. Nucleic Acids Res. 2015. PMID: 25820428 Free PMC article.
-
Next generation sequencing in cancer research and clinical application.Biol Proced Online. 2013 Feb 13;15(1):4. doi: 10.1186/1480-9222-15-4. Biol Proced Online. 2013. PMID: 23406336 Free PMC article.
-
PrfA-like transcription factor gene lmo0753 contributes to L-rhamnose utilization in Listeria monocytogenes strains associated with human food-borne infections.Appl Environ Microbiol. 2013 Sep;79(18):5584-92. doi: 10.1128/AEM.01812-13. Epub 2013 Jul 8. Appl Environ Microbiol. 2013. PMID: 23835178 Free PMC article.
-
Brain Transcriptomics of Wild and Domestic Rabbits Suggests That Changes in Dopamine Signaling and Ciliary Function Contributed to Evolution of Tameness.Genome Biol Evol. 2020 Oct 1;12(10):1918-1928. doi: 10.1093/gbe/evaa158. Genome Biol Evol. 2020. PMID: 32835359 Free PMC article.
-
Genome sequencing and mapping reveal loss of heterozygosity as a mechanism for rapid adaptation in the vegetable pathogen Phytophthora capsici.Mol Plant Microbe Interact. 2012 Oct;25(10):1350-60. doi: 10.1094/MPMI-02-12-0028-R. Mol Plant Microbe Interact. 2012. PMID: 22712506 Free PMC article.
References
-
- Bhangale TR, et al. Comprehensive identification and characterization of diallelic insertion-deletion polymorphisms in 330 human candidate genes. Hum. Mol. Genet. 2005;14:59–69. - PubMed
-
- Bona FD, et al. Optimal spliced alignments of short sequence reads. Bioinformatics. 2008;24:i174–180. - PubMed
-
- Burrows M, Wheeler DJ. Technical Report 124. California: Digital Equipment Corporation, Palo Alto; 1994. A block-sorting lossless data compression algorithm.
-
- Canales RD, et al. Evaluation of DNA microarray results with quantitative gene expression platforms. Nat. Biotechnol. 2006;24:1115–1122. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
