

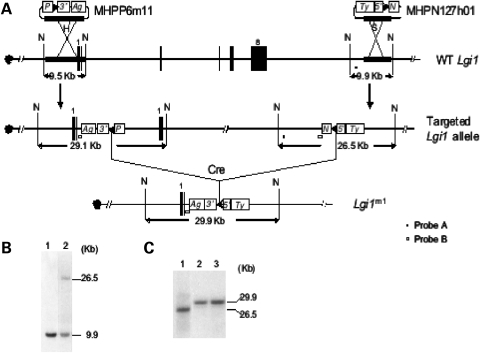
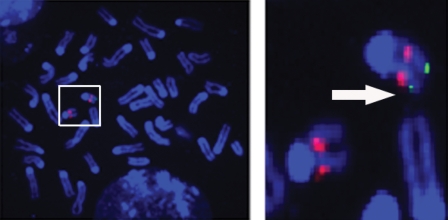
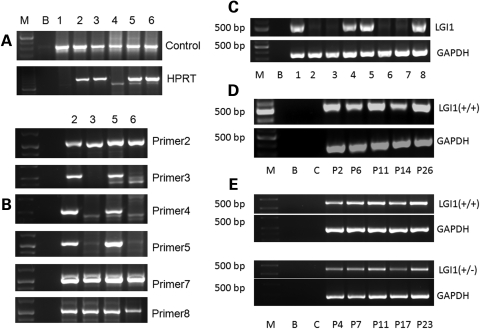
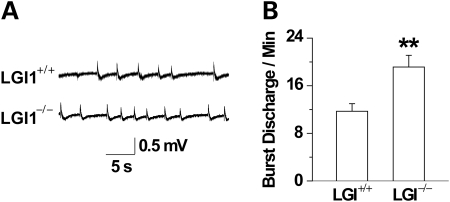
Lgi1 null mutant mice exhibit myoclonic seizures and CA1 neuronal hyperexcitability
- PMID: 20130004
- PMCID: PMC2850618
- DOI: 10.1093/hmg/ddq047
Lgi1 null mutant mice exhibit myoclonic seizures and CA1 neuronal hyperexcitability
Abstract
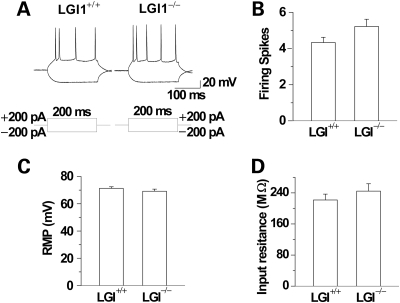
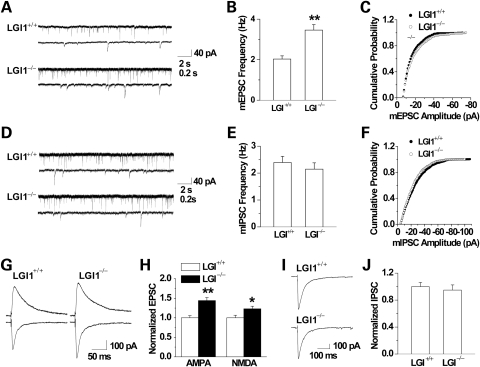
LGI1 in humans is responsible for a predisposition to autosomal dominant partial epilepsy with auditory features (ADPEAF). However, mechanisms of how LGI1 mutations cause epilepsy remain unclear. We have used a mouse chromosome engineering strategy to create a null mutation for the gene ortholog encoding LGI1. The Lgi1 null mutant mice show no gross overall developmental abnormalities from routine histopathological analysis. After 12-18 days of age, the homozygous mutant mice all exhibit myoclonic seizures accompanied by rapid jumping and running and die shortly thereafter. The heterozygous mutant mice do not develop seizures. Electrophysiological analysis demonstrates an enhanced excitatory synaptic transmission by increasing the release of the excitatory neurotransmitter glutamate, suggesting a basis for the seizure phenotype. This mouse model, therefore, provides novel insights into the mechanism behind ADPEAF and offers a new opportunity to study the mechanism behind the role of LGI1 in susceptibility to myoclonic seizures.
Figures
Similar articles
-
Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy.Proc Natl Acad Sci U S A. 2010 Feb 23;107(8):3799-804. doi: 10.1073/pnas.0914537107. Epub 2010 Feb 2. Proc Natl Acad Sci U S A. 2010. PMID: 20133599 Free PMC article.
-
A rat model for LGI1-related epilepsies.Hum Mol Genet. 2012 Aug 15;21(16):3546-57. doi: 10.1093/hmg/dds184. Epub 2012 May 15. Hum Mol Genet. 2012. PMID: 22589250
-
Electroclinical characterization of epileptic seizures in leucine-rich, glioma-inactivated 1-deficient mice.Brain. 2010 Sep;133(9):2749-62. doi: 10.1093/brain/awq171. Epub 2010 Jul 21. Brain. 2010. PMID: 20659958 Free PMC article.
-
LGI1: from zebrafish to human epilepsy.Prog Brain Res. 2014;213:159-79. doi: 10.1016/B978-0-444-63326-2.00009-0. Prog Brain Res. 2014. PMID: 25194489 Review.
-
LGI1 mutations in autosomal dominant and sporadic lateral temporal epilepsy.Hum Mutat. 2009 Apr;30(4):530-6. doi: 10.1002/humu.20925. Hum Mutat. 2009. PMID: 19191227 Review.
Cited by
-
Similarity of molecular phenotype between known epilepsy gene LGI1 and disease candidate gene LGI2.BMC Biochem. 2010 Sep 24;11:39. doi: 10.1186/1471-2091-11-39. BMC Biochem. 2010. PMID: 20863412 Free PMC article.
-
The very large G protein coupled receptor (Vlgr1) in hair cells.J Mol Neurosci. 2013 May;50(1):204-14. doi: 10.1007/s12031-012-9911-5. Epub 2012 Nov 20. J Mol Neurosci. 2013. PMID: 23180093 Review.
-
Essential roles of leucine-rich glioma inactivated 1 in the development of embryonic and postnatal cerebellum.Sci Rep. 2015 Jan 16;5:7827. doi: 10.1038/srep07827. Sci Rep. 2015. PMID: 25591666 Free PMC article.
-
Case Report: Three Case Reports of Rapidly Progressive Dementias and Narrative Review.Case Rep Neurol. 2022 Nov 11;14(3):441-455. doi: 10.1159/000525701. eCollection 2022 Sep-Dec. Case Rep Neurol. 2022. PMID: 36636277 Free PMC article.
-
Clinical spectrum and diagnostic value of antibodies against the potassium channel related protein complex.Neurologia. 2015 Jun;30(5):295-301. doi: 10.1016/j.nrl.2013.12.007. Epub 2014 Jan 30. Neurologia. 2015. PMID: 24485651 Free PMC article. Review.
References
-
- Michelucci R., Poza J.J., Sofia V., de Feo M.R., Binelli S., Bisulli F., Scudellaro E., Simionati B., Zimbello R., D'Orsi G., et al. Autosomal dominant lateral temporal epilepsy: clinical spectrum, new epitempin mutations, and genetic heterogeneity in seven European families. Epilepsia. 2003;44:1289–1297. - PubMed
-
- Poza J.J., Sáenz A., Martínez-Gil A., Cheron N., Cobo A.M., Urtasun M., Martí-Massó J.F., Grid D., Beckmann J.S., Prud'homme J.F., et al. Autosomal dominant lateral temporal epilepsy: clinical and genetic study of a large Basque pedigree linked to chromosome 10q. Ann. Neurol. 1999;45:182–188. - PubMed
-
- Morante-Redolate J.M., Gorostidi-Pagola A., Piquer-Sirerol S., Saenz A., Poza J.R., Galan J., Gesk S., Sarafidou T., Mautner V.F., Binelli S., et al. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temportal epilepsy. Hum. Mol. Genet. 2002;11:1119–1128. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
