

Wild-type p53-induced phosphatase 1 dephosphorylates histone variant gamma-H2AX and suppresses DNA double strand break repair
- PMID: 20118229
- PMCID: PMC2857113
- DOI: 10.1074/jbc.M109.071696
Wild-type p53-induced phosphatase 1 dephosphorylates histone variant gamma-H2AX and suppresses DNA double strand break repair
Abstract
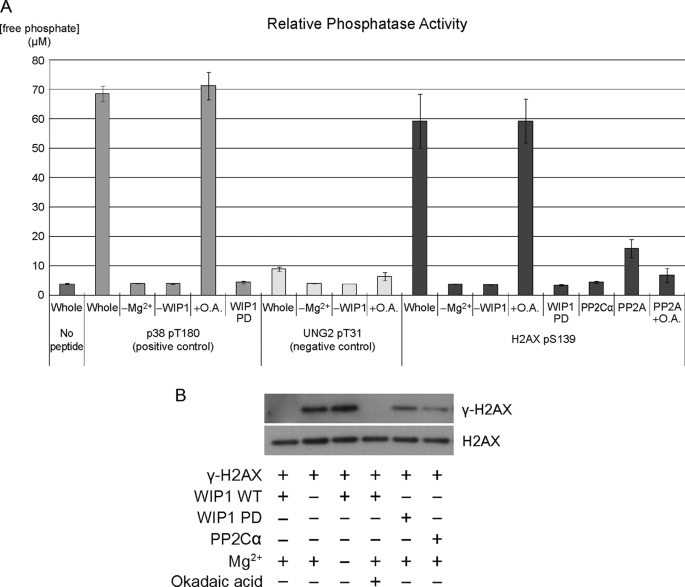
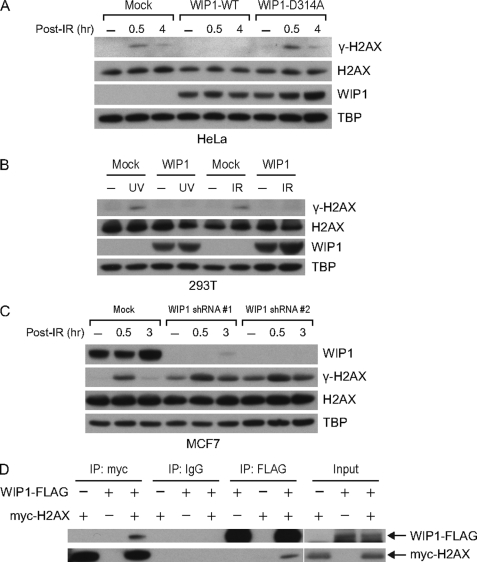
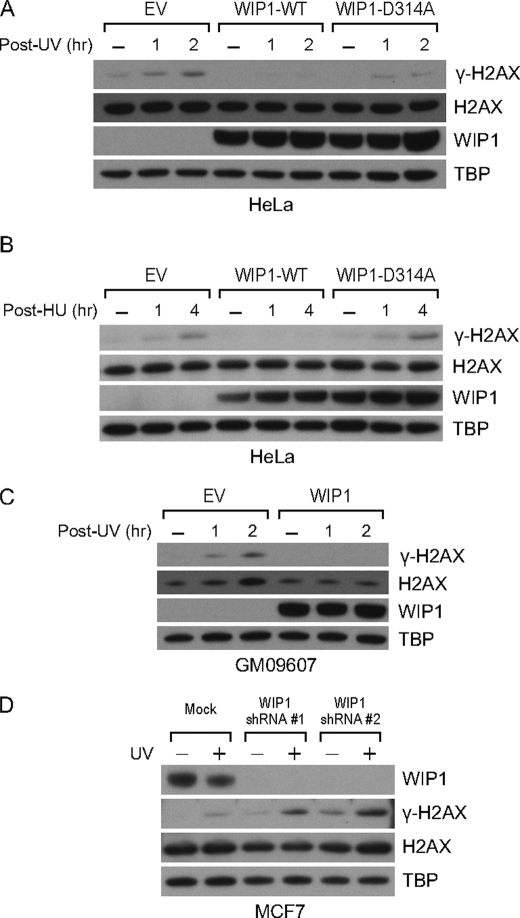
In response to DNA double strand breaks, the histone variant H2AX at the break site is phosphorylated at serine 139 by DNA damage sensor kinases such as ataxia telangiectasia-mutated, forming gamma-H2AX. This phosphorylation event is critical for sustained recruitment of other proteins to repair the break. After repair, restoration of the cell to a prestress state is associated with gamma-H2AX dephosphorylation and dissolution of gamma-H2AX-associated damage foci. The phosphatases PP2A and PP4 have previously been shown to dephosphorylate gamma-H2AX. Here, we demonstrate that the wild-type p53-induced phosphatase 1 (WIP1) also dephosphorylates gamma-H2AX at serine 139 in vitro and in vivo. Overexpression of WIP1 reduces formation of gamma-H2AX foci in response to ionizing and ultraviolet radiation and blocks recruitment of MDC1 (mediator of DNA damage checkpoint 1) and 53BP1 (p53 binding protein 1) to DNA damage foci. Finally, these inhibitory effects of WIP1 on gamma-H2AX are accompanied by WIP1 suppression of DNA double strand break repair. Thus, WIP1 has a homeostatic role in reversing the effects of ataxia telangiectasia-mutated phosphorylation of H2AX.
Figures





Similar articles
-
Dephosphorylation of γ-H2AX by WIP1: an important homeostatic regulatory event in DNA repair and cell cycle control.Cell Cycle. 2010 Jun 1;9(11):2092-6. doi: 10.4161/cc.9.11.11810. Epub 2010 Jun 1. Cell Cycle. 2010. PMID: 20495376 Free PMC article.
-
Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates gamma-H2AX.Mol Cell Biol. 2010 Mar;30(6):1368-81. doi: 10.1128/MCB.00741-09. Epub 2010 Jan 11. Mol Cell Biol. 2010. PMID: 20065038 Free PMC article.
-
Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells.Proc Natl Acad Sci U S A. 2011 Sep 6;108(36):14944-9. doi: 10.1073/pnas.1100959108. Epub 2011 Sep 6. Proc Natl Acad Sci U S A. 2011. PMID: 21896770 Free PMC article.
-
Laser-induced radiation microbeam technology and simultaneous real-time fluorescence imaging in live cells.Methods Enzymol. 2012;504:3-28. doi: 10.1016/B978-0-12-391857-4.00001-X. Methods Enzymol. 2012. PMID: 22264527 Review.
-
The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax.DNA Repair (Amst). 2010 Dec 10;9(12):1273-82. doi: 10.1016/j.dnarep.2010.09.013. Epub 2010 Oct 30. DNA Repair (Amst). 2010. PMID: 21036673 Review.
Cited by
-
Wip1 suppresses apoptotic cell death through direct dephosphorylation of BAX in response to γ-radiation.Cell Death Dis. 2013 Aug 1;4(8):e744. doi: 10.1038/cddis.2013.252. Cell Death Dis. 2013. PMID: 23907458 Free PMC article.
-
Analysis of miRNA and mRNA expression profiles highlights alterations in ionizing radiation response of human lymphocytes under modeled microgravity.PLoS One. 2012;7(2):e31293. doi: 10.1371/journal.pone.0031293. Epub 2012 Feb 9. PLoS One. 2012. PMID: 22347458 Free PMC article.
-
Inhibition of mutant PPM1D enhances DNA damage response and growth suppressive effects of ionizing radiation in diffuse intrinsic pontine glioma.Neuro Oncol. 2019 Jun 10;21(6):786-799. doi: 10.1093/neuonc/noz053. Neuro Oncol. 2019. PMID: 30852603 Free PMC article.
-
Ionizing radiation-induced responses in human cells with differing TP53 status.Int J Mol Sci. 2013 Nov 13;14(11):22409-35. doi: 10.3390/ijms141122409. Int J Mol Sci. 2013. PMID: 24232458 Free PMC article. Review.
-
Absence of Wip1 partially rescues Atm deficiency phenotypes in mice.Oncogene. 2012 Mar 1;31(9):1155-65. doi: 10.1038/onc.2011.303. Epub 2011 Jul 18. Oncogene. 2012. PMID: 21765465 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
