

The implications of patterns in HIV diversity for neutralizing antibody induction and susceptibility
- PMID: 20048705
- PMCID: PMC6426297
- DOI: 10.1097/COH.0b013e32832f129e
The implications of patterns in HIV diversity for neutralizing antibody induction and susceptibility
Abstract
Purpose of review: Designing an HIV vaccine capable of eliciting broadly cross-reactive neutralizing antibodies is an extraordinarily difficult challenge. Here we focus on the implications of HIV diversity for vaccine design, detailing the impact of levels of variation in epitopes of known potent neutralizing antibodies, and summarizing patterns of overall variation in regional domains within gp120. Strategies for rational vaccine design, to enhance coverage of HIV's natural diversity, are considered.
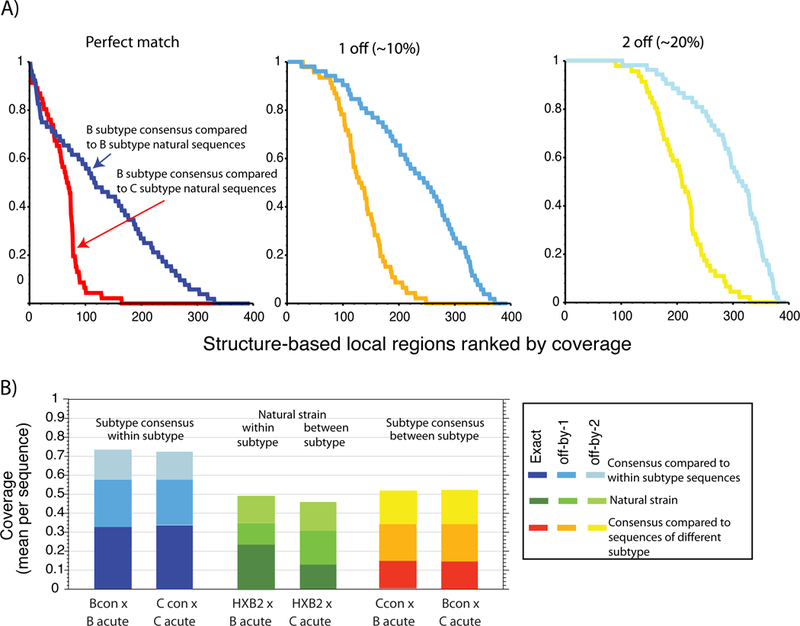
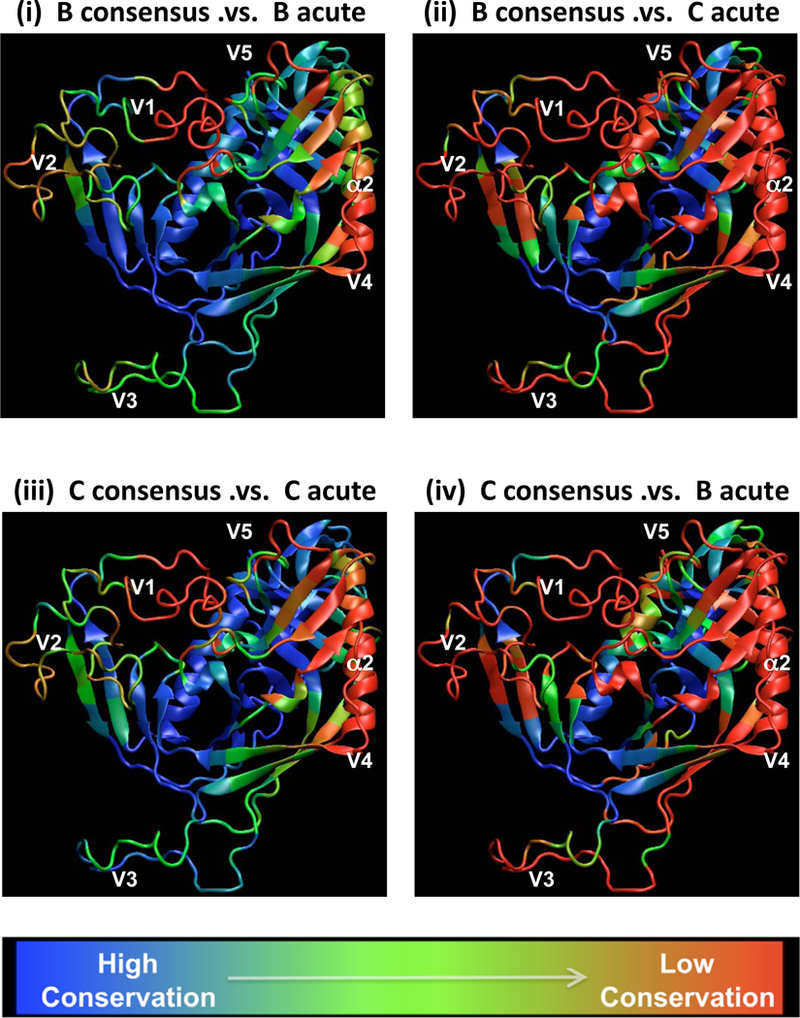
Recent findings: Each amino acid in an envelope gp120 three-dimensional structure was grouped with its 10 nearest neighbors and classified by their natural sequence variability. Within-subtype variation is superimposed on patterns of subtype-specific variation. Regions under selection with moderate diversity are realistic vaccine targets; their variation reflects the value of escape in these regions, whereas the level of diversity is potentially approachable with a vaccine.
Summary: HIV diversity is so extensive that vaccine design strategies may benefit by factoring in diversity from the earliest stages, even for vaccines that target relatively conserved regions.
Figures

Similar articles
-
A Trimeric HIV-1 Envelope gp120 Immunogen Induces Potent and Broad Anti-V1V2 Loop Antibodies against HIV-1 in Rabbits and Rhesus Macaques.J Virol. 2018 Feb 12;92(5):e01796-17. doi: 10.1128/JVI.01796-17. Print 2018 Mar 1. J Virol. 2018. PMID: 29237847 Free PMC article.
-
Conformational Epitope-Specific Broadly Neutralizing Plasma Antibodies Obtained from an HIV-1 Clade C-Infected Elite Neutralizer Mediate Autologous Virus Escape through Mutations in the V1 Loop.J Virol. 2016 Jan 13;90(7):3446-57. doi: 10.1128/JVI.03090-15. J Virol. 2016. PMID: 26763999 Free PMC article.
-
GP120: target for neutralizing HIV-1 antibodies.Annu Rev Immunol. 2006;24:739-69. doi: 10.1146/annurev.immunol.24.021605.090557. Annu Rev Immunol. 2006. PMID: 16551265 Review.
-
Bacterially expressed HIV-1 gp120 outer-domain fragment immunogens with improved stability and affinity for CD4-binding site neutralizing antibodies.J Biol Chem. 2018 Sep 28;293(39):15002-15020. doi: 10.1074/jbc.RA118.005006. Epub 2018 Aug 9. J Biol Chem. 2018. PMID: 30093409 Free PMC article.
-
Structure-function relationships of HIV-1 envelope sequence-variable regions refocus vaccine design.Nat Rev Immunol. 2010 Jul;10(7):527-35. doi: 10.1038/nri2801. Nat Rev Immunol. 2010. PMID: 20577269 Free PMC article. Review.
Cited by
-
Viral CTL escape mutants are generated in lymph nodes and subsequently become fixed in plasma and rectal mucosa during acute SIV infection of macaques.PLoS Pathog. 2011 May;7(5):e1002048. doi: 10.1371/journal.ppat.1002048. Epub 2011 May 19. PLoS Pathog. 2011. PMID: 21625590 Free PMC article.
-
Impact of HIV-1 Diversity on Its Sensitivity to Neutralization.Vaccines (Basel). 2019 Jul 25;7(3):74. doi: 10.3390/vaccines7030074. Vaccines (Basel). 2019. PMID: 31349655 Free PMC article. Review.
-
Strategies for inducing effective neutralizing antibody responses against HIV-1.Expert Rev Vaccines. 2019 Nov;18(11):1127-1143. doi: 10.1080/14760584.2019.1690458. Epub 2019 Dec 2. Expert Rev Vaccines. 2019. PMID: 31791150 Free PMC article. Review.
-
Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1.Science. 2010 Aug 13;329(5993):856-61. doi: 10.1126/science.1187659. Epub 2010 Jul 8. Science. 2010. PMID: 20616233 Free PMC article.
-
HIV control is mediated in part by CD8+ T-cell targeting of specific epitopes.J Virol. 2014 Nov;88(22):12937-48. doi: 10.1128/JVI.01004-14. Epub 2014 Aug 27. J Virol. 2014. PMID: 25165115 Free PMC article.
References
-
- Eckert DM, Kim PS: Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem 2001, 70:777–810 - PubMed
-
- Wyatt R, Sodroski J: The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 1998, 280:1884–8 - PubMed
-
- Wyatt R, Kwong PD, Desjardins E et al.: The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 1998, 393:705–11 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
