

Protein kinase C zeta mediates cigarette smoke/aldehyde- and lipopolysaccharide-induced lung inflammation and histone modifications
- PMID: 20007975
- PMCID: PMC2820769
- DOI: 10.1074/jbc.M109.041418
Protein kinase C zeta mediates cigarette smoke/aldehyde- and lipopolysaccharide-induced lung inflammation and histone modifications
Abstract
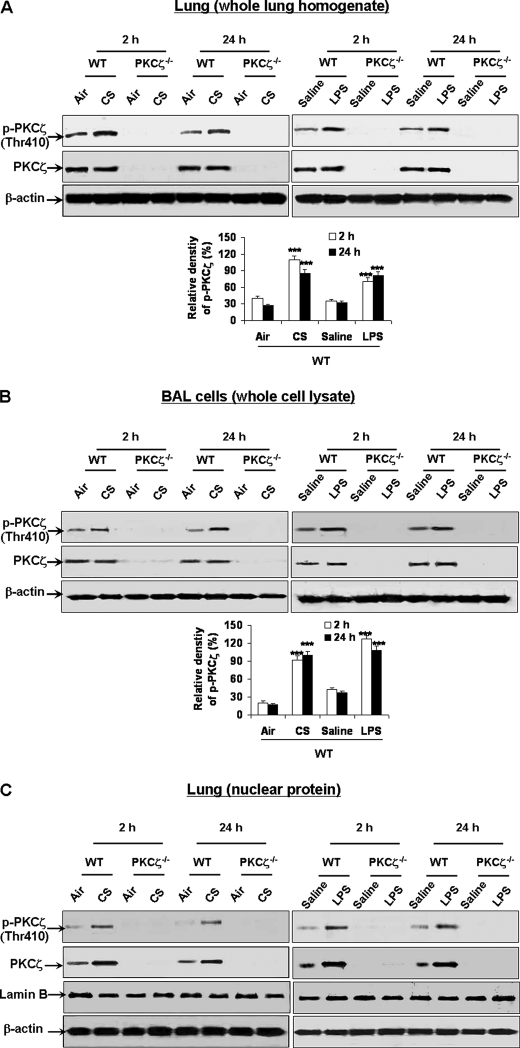
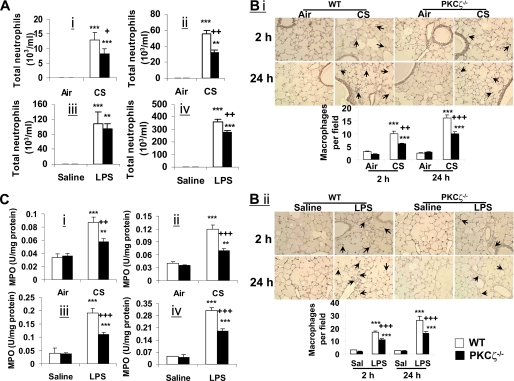
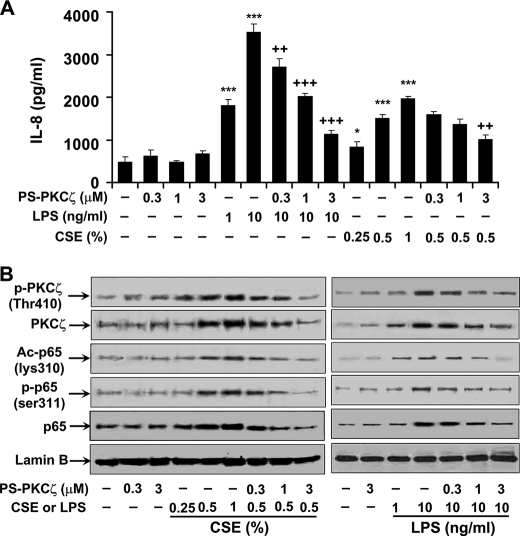
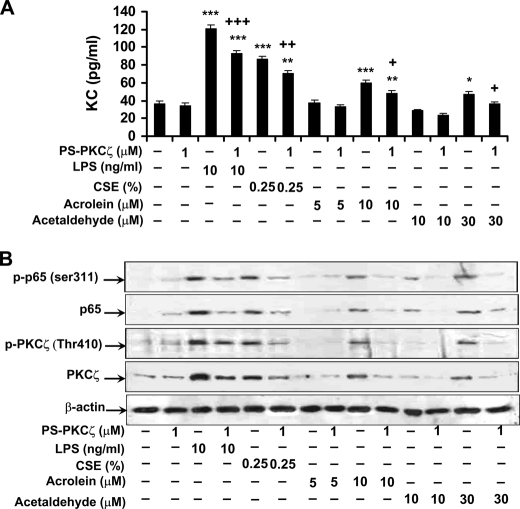
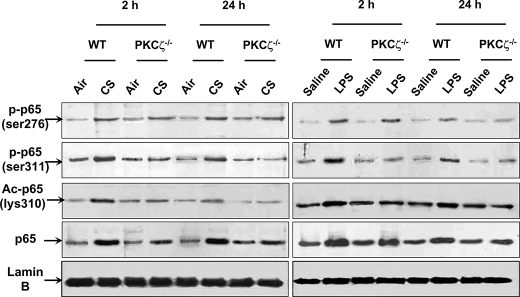
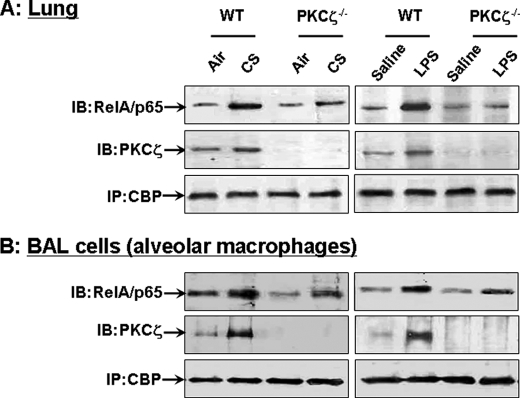
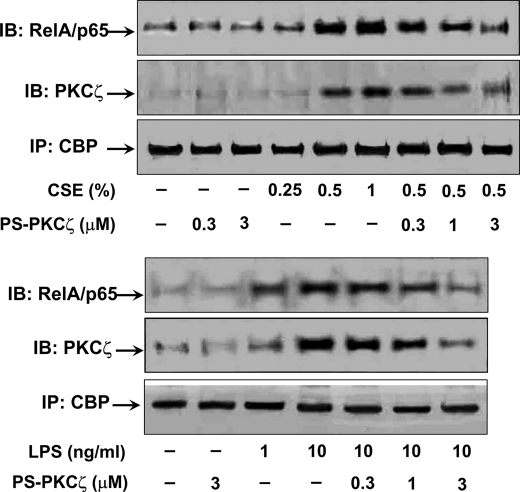
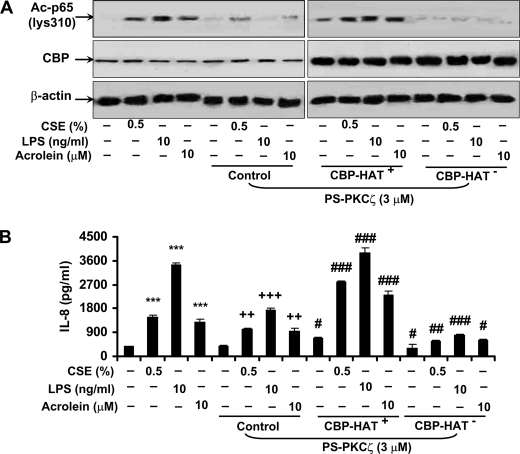
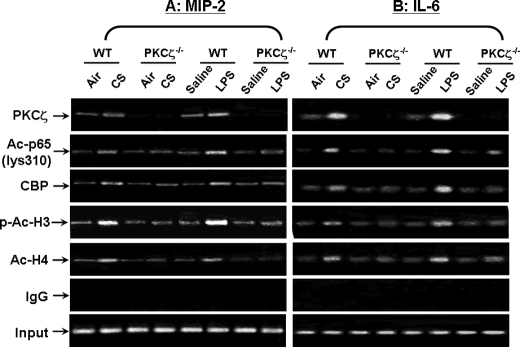
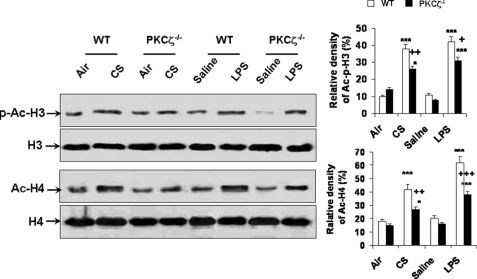
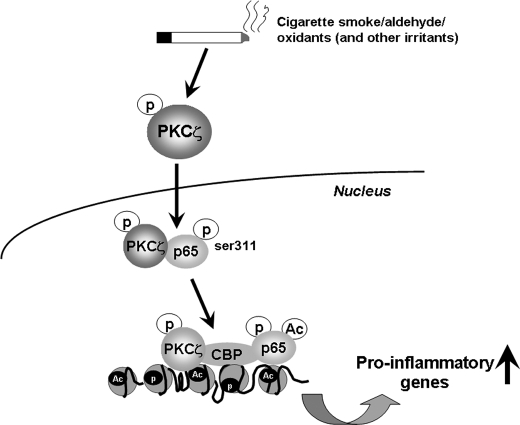
Atypical protein kinase C (PKC) zeta is an important regulator of inflammation through activation of the nuclear factor-kappaB (NF-kappaB) pathway. Chromatin remodeling on pro-inflammatory genes plays a pivotal role in cigarette smoke (CS)- and lipopolysaccharide (LPS)-induced abnormal lung inflammation. However, the signaling mechanism whereby chromatin remodeling occurs in CS- and LPS-induced lung inflammation is not known. We hypothesized that PKCzeta is an important regulator of chromatin remodeling, and down-regulation of PKCzeta ameliorates lung inflammation by CS and LPS exposures. We determined the role and molecular mechanism of PKCzeta in abnormal lung inflammatory response to CS and LPS exposures in PKCzeta-deficient (PKCzeta(-/-)) and wild-type mice. Lung inflammatory response was decreased in PKCzeta(-/-) mice compared with WT mice exposed to CS and LPS. Moreover, inhibition of PKCzeta by a specific pharmacological PKCzeta inhibitor attenuated CS extract-, reactive aldehydes (present in CS)-, and LPS-mediated pro-inflammatory mediator release from macrophages. The mechanism underlying these findings is associated with decreased RelA/p65 phosphorylation (Ser(311)) and translocation of the RelA/p65 subunit of NF-kappaB into the nucleus. Furthermore, CS/reactive aldehydes and LPS exposures led to activation and translocation of PKCzeta into the nucleus where it forms a complex with CREB-binding protein (CBP) and acetylated RelA/p65 causing histone phosphorylation and acetylation on promoters of pro-inflammatory genes. Taken together, these data suggest that PKCzeta plays an important role in CS/aldehyde- and LPS-induced lung inflammation through acetylation of RelA/p65 and histone modifications via CBP. These data provide new insights into the molecular mechanisms underlying the pathogenesis of chronic inflammatory lung diseases.
Figures
Similar articles
-
Mitogen- and stress-activated kinase 1 (MSK1) regulates cigarette smoke-induced histone modifications on NF-κB-dependent genes.PLoS One. 2012;7(2):e31378. doi: 10.1371/journal.pone.0031378. Epub 2012 Feb 1. PLoS One. 2012. PMID: 22312446 Free PMC article.
-
Targeted disruption of NF-{kappa}B1 (p50) augments cigarette smoke-induced lung inflammation and emphysema in mice: a critical role of p50 in chromatin remodeling.Am J Physiol Lung Cell Mol Physiol. 2010 Feb;298(2):L197-209. doi: 10.1152/ajplung.00265.2009. Epub 2009 Dec 4. Am J Physiol Lung Cell Mol Physiol. 2010. PMID: 19965984 Free PMC article.
-
IKK alpha causes chromatin modification on pro-inflammatory genes by cigarette smoke in mouse lung.Am J Respir Cell Mol Biol. 2008 Jun;38(6):689-98. doi: 10.1165/rcmb.2007-0379OC. Epub 2008 Jan 31. Am J Respir Cell Mol Biol. 2008. PMID: 18239189 Free PMC article.
-
Oxidative stress and chromatin remodeling in chronic obstructive pulmonary disease and smoking-related diseases.Antioxid Redox Signal. 2013 May 20;18(15):1956-71. doi: 10.1089/ars.2012.4863. Epub 2012 Nov 6. Antioxid Redox Signal. 2013. PMID: 22978694 Free PMC article. Review.
-
Redox regulation of SIRT1 in inflammation and cellular senescence.Free Radic Biol Med. 2013 Aug;61:95-110. doi: 10.1016/j.freeradbiomed.2013.03.015. Epub 2013 Mar 27. Free Radic Biol Med. 2013. PMID: 23542362 Free PMC article. Review.
Cited by
-
NF-κB inducing kinase, NIK mediates cigarette smoke/TNFα-induced histone acetylation and inflammation through differential activation of IKKs.PLoS One. 2011;6(8):e23488. doi: 10.1371/journal.pone.0023488. Epub 2011 Aug 24. PLoS One. 2011. PMID: 21887257 Free PMC article.
-
Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary disease.FASEB J. 2015 Jul;29(7):2912-29. doi: 10.1096/fj.14-268276. Epub 2015 Mar 19. FASEB J. 2015. PMID: 25792665 Free PMC article.
-
Airway inflammation and hypersensitivity induced by chronic smoking.Respir Physiol Neurobiol. 2011 Sep 30;178(3):395-405. doi: 10.1016/j.resp.2011.03.004. Epub 2011 Mar 10. Respir Physiol Neurobiol. 2011. PMID: 21397052 Free PMC article. Review.
-
Cigarette smoke induces distinct histone modifications in lung cells: implications for the pathogenesis of COPD and lung cancer.J Proteome Res. 2014 Feb 7;13(2):982-96. doi: 10.1021/pr400998n. Epub 2013 Dec 13. J Proteome Res. 2014. PMID: 24283195 Free PMC article.
-
KLF6 and iNOS regulates apoptosis during respiratory syncytial virus infection.Cell Immunol. 2013 May-Jun;283(1-2):1-7. doi: 10.1016/j.cellimm.2013.06.002. Epub 2013 Jun 18. Cell Immunol. 2013. PMID: 23831683 Free PMC article.
References
-
- Yang S. R., Wright J., Bauter M., Seweryniak K., Kode A., Rahman I. (2007) Am. J. Physiol. Lung Cell Mol. Physiol. 292, L567–L576 - PubMed
-
- Yao H., Edirisinghe I., Rajendrasozhan S., Yang S. R., Caito S., Adenuga D., Rahman I. (2008) Am. J. Physiol. Lung Cell Mol. Physiol. 294, L1174–L1186 - PubMed
-
- Hogg J. C., Chu F., Utokaparch S., Woods R., Elliott W. M., Buzatu L., Cherniack R. M., Rogers R. M., Sciurba F. C., Coxson H. O., Paré P. D. (2004) N. Engl. J. Med. 350, 2645–2653 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
