

The conformational transition pathway of ATP binding cassette transporter MsbA revealed by atomistic simulations
- PMID: 19996093
- PMCID: PMC2823423
- DOI: 10.1074/jbc.M109.056432
The conformational transition pathway of ATP binding cassette transporter MsbA revealed by atomistic simulations
Abstract
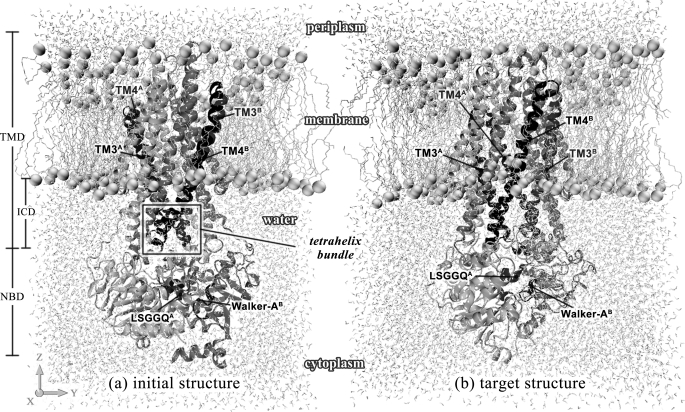
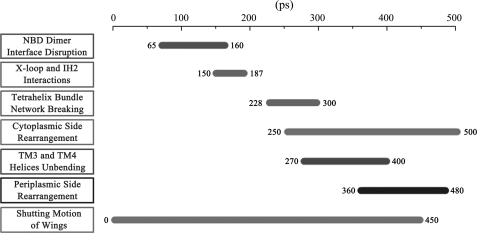
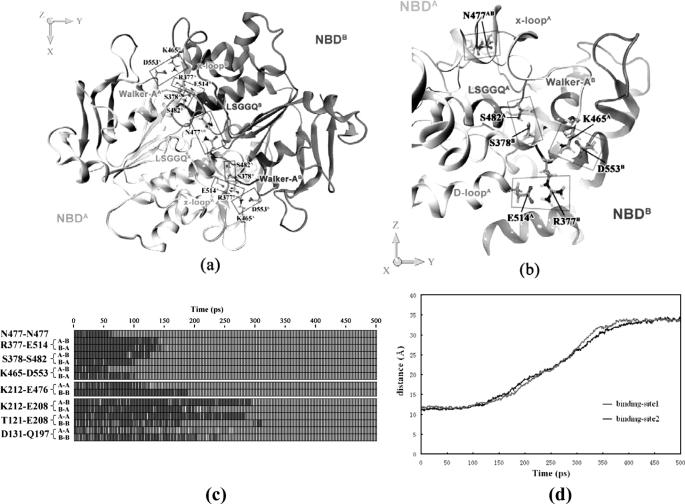
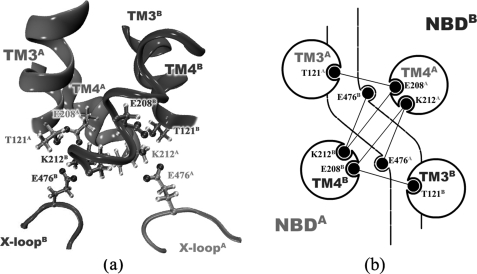
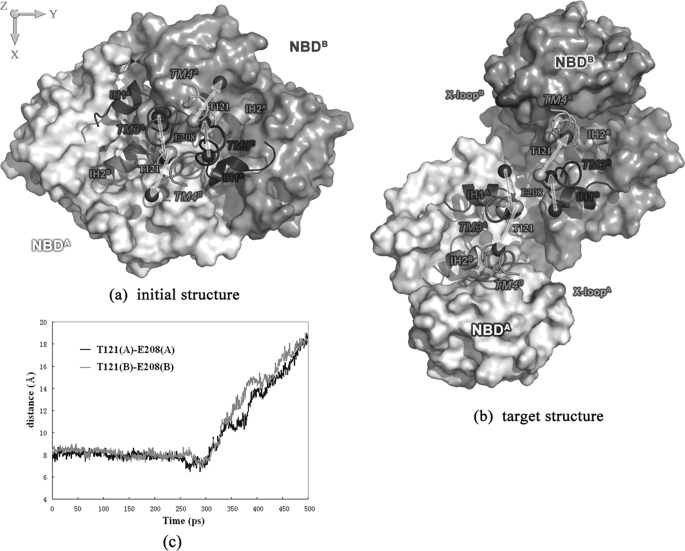
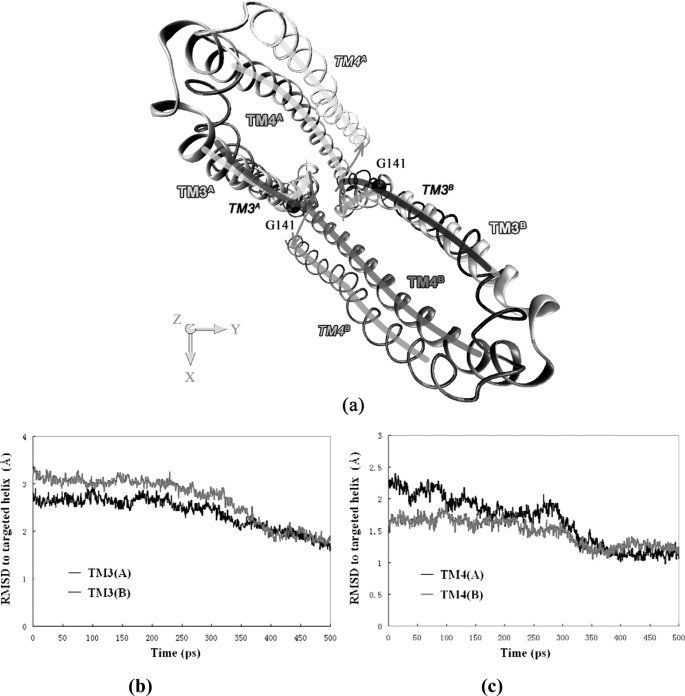
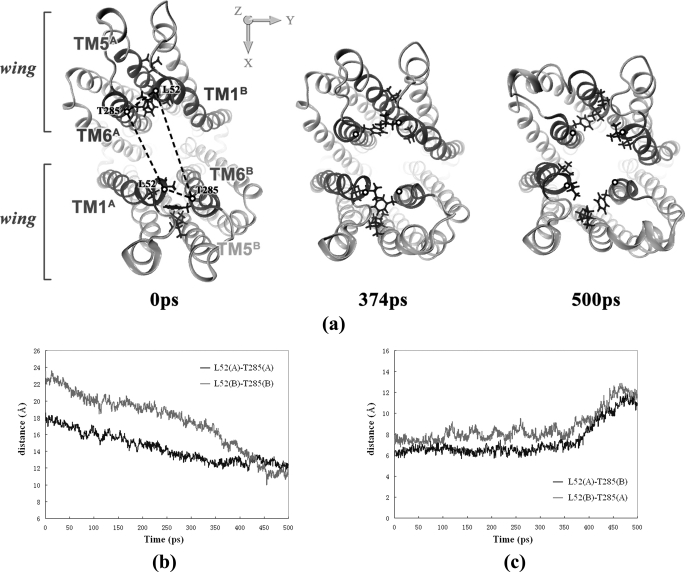
ATP binding cassette transporters are integral membrane proteins that use the energy released from ATP hydrolysis at the two nucleotide binding domains (NBDs) to translocate a wide variety of substrates through a channel at the two transmembrane domains (TMDs) across the cell membranes. MsbA from Gram-negative bacteria is a lipid and multidrug resistance ATP binding cassette exporter that can undergo large scale conformational changes between the outward-facing and the inward-facing conformations revealed by crystal structures in different states. Here, we use targeted molecular dynamics simulation methods to explore the atomic details of the conformational transition from the outward-facing to the inward-facing states of MsbA. The molecular dynamics trajectories revealed a clear spatiotemporal order of the conformational movements. The disruption of the nucleotide binding sites at the NBD dimer interface is the very first event that initiates the following conformational changes, verifying the assumption that the conformational conversion is triggered by ATP hydrolysis. The conserved x-loops of the NBDs were identified to participate in the interaction network that stabilizes the cytoplasmic tetrahelix bundle of the TMDs and play an important role in mediating the cross-talk between the NBD and TMD. The movement of the NBD dimer is transmitted through x-loops to break the tetrahelix bundle, inducing the packing rearrangements of the transmembrane helices at the cytoplasmic side and the periplasmic side sequentially. The packing rearrangement within each periplasmic wing of TMD that results in exposure of the substrate binding sites occurred at the end stage of the trajectory, preventing the wrong timing of the binding site accessibility.
Figures
Similar articles
-
Molecular disruption of the power stroke in the ATP-binding cassette transport protein MsbA.J Biol Chem. 2013 Mar 8;288(10):6801-13. doi: 10.1074/jbc.M112.430074. Epub 2013 Jan 10. J Biol Chem. 2013. PMID: 23306205 Free PMC article.
-
The conformational transition pathways of ATP-binding cassette transporter BtuCD revealed by targeted molecular dynamics simulation.PLoS One. 2012;7(1):e30465. doi: 10.1371/journal.pone.0030465. Epub 2012 Jan 17. PLoS One. 2012. PMID: 22272354 Free PMC article.
-
The Lipid Bilayer Modulates the Structure and Function of an ATP-binding Cassette Exporter.J Biol Chem. 2016 Feb 26;291(9):4453-61. doi: 10.1074/jbc.M115.698498. Epub 2016 Jan 2. J Biol Chem. 2016. PMID: 26725230 Free PMC article.
-
Structural dynamics of ABC transporters: molecular simulation studies.Biochem Soc Trans. 2021 Feb 26;49(1):405-414. doi: 10.1042/BST20200710. Biochem Soc Trans. 2021. PMID: 33634827 Review.
-
What monomeric nucleotide binding domains can teach us about dimeric ABC proteins.FEBS Lett. 2020 Dec;594(23):3857-3875. doi: 10.1002/1873-3468.13921. Epub 2020 Sep 17. FEBS Lett. 2020. PMID: 32880928 Review.
Cited by
-
The high-energy transition state of the glutamate transporter homologue GltPh.EMBO J. 2021 Jan 4;40(1):e105415. doi: 10.15252/embj.2020105415. Epub 2020 Nov 13. EMBO J. 2021. PMID: 33185289 Free PMC article.
-
In silico model for P-glycoprotein substrate prediction: insights from molecular dynamics and in vitro studies.J Comput Aided Mol Des. 2013 Apr;27(4):347-63. doi: 10.1007/s10822-013-9650-x. Epub 2013 Apr 24. J Comput Aided Mol Des. 2013. PMID: 23612916
-
G551D mutation impairs PKA-dependent activation of CFTR channel that can be restored by novel GOF mutations.Am J Physiol Lung Cell Mol Physiol. 2020 Nov 1;319(5):L770-L785. doi: 10.1152/ajplung.00262.2019. Epub 2020 Sep 2. Am J Physiol Lung Cell Mol Physiol. 2020. PMID: 32877225 Free PMC article.
-
Inter-domain communication mechanisms in an ABC importer: a molecular dynamics study of the MalFGK2E complex.PLoS Comput Biol. 2011 Aug;7(8):e1002128. doi: 10.1371/journal.pcbi.1002128. Epub 2011 Aug 4. PLoS Comput Biol. 2011. PMID: 21829343 Free PMC article.
-
Substrate binding accelerates the conformational transitions and substrate dissociation in multidrug efflux transporter AcrB.Front Microbiol. 2015 Apr 13;6:302. doi: 10.3389/fmicb.2015.00302. eCollection 2015. Front Microbiol. 2015. PMID: 25918513 Free PMC article.
References
-
- Dean M., Allikmets R. (2001) J. Bioenerg. Biomembr. 33, 475–479 - PubMed
-
- Hughes A. L. (1994) Mol. Biol. Evol. 11, 899–910 - PubMed
-
- Ling V. (1992) Cancer 69, 2603–2609 - PubMed
-
- Raviv Y., Pollard H. B., Bruggemann E. P., Pastan I., Gottesman M. M. (1990) J. Biol. Chem. 265, 3975–3980 - PubMed
-
- Davidson A. L., Chen J. (2004) Annu. Rev. Biochem. 73, 241–268 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
