

Mitochondria, bioenergetics, and the epigenome in eukaryotic and human evolution
- PMID: 19955254
- PMCID: PMC3905750
- DOI: 10.1101/sqb.2009.74.031
Mitochondria, bioenergetics, and the epigenome in eukaryotic and human evolution
Abstract
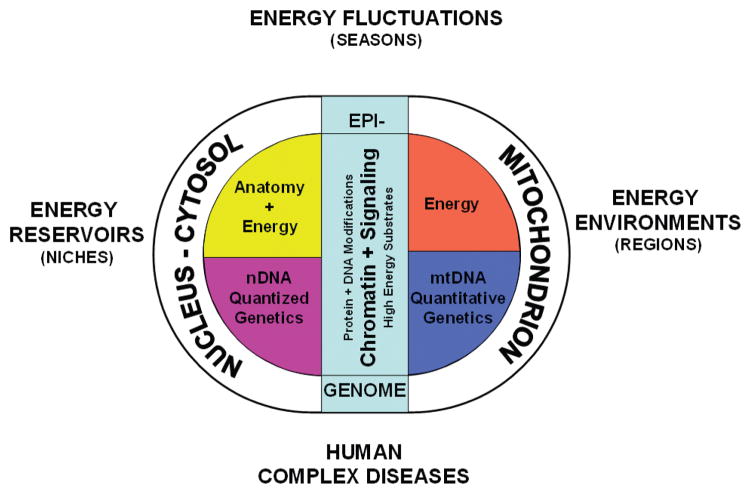
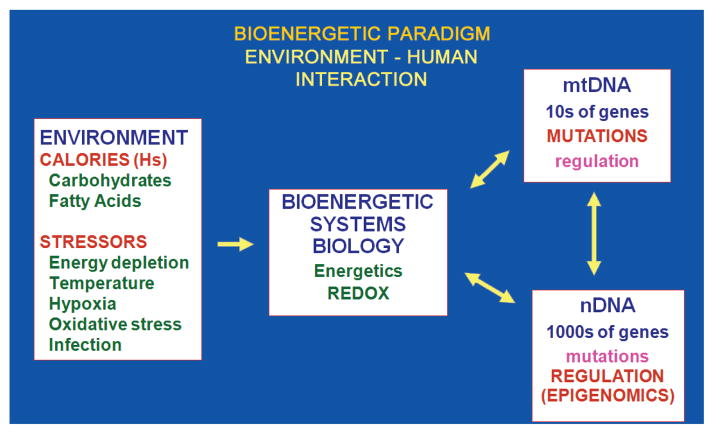
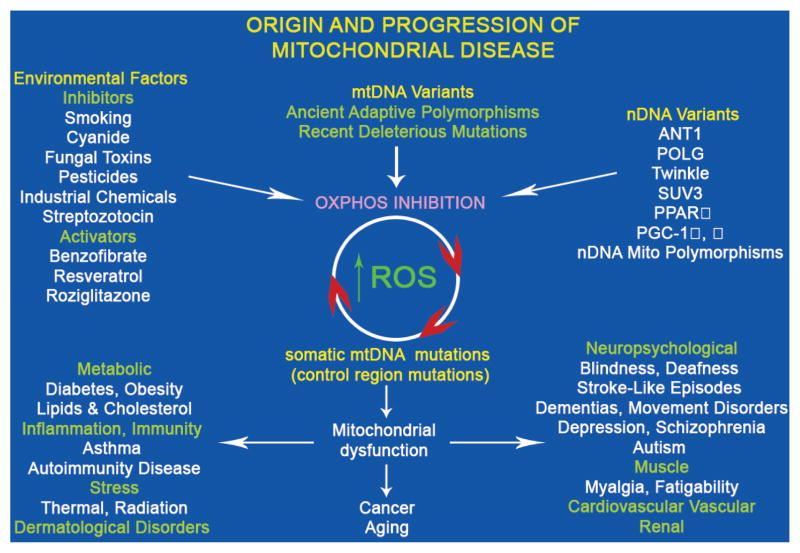
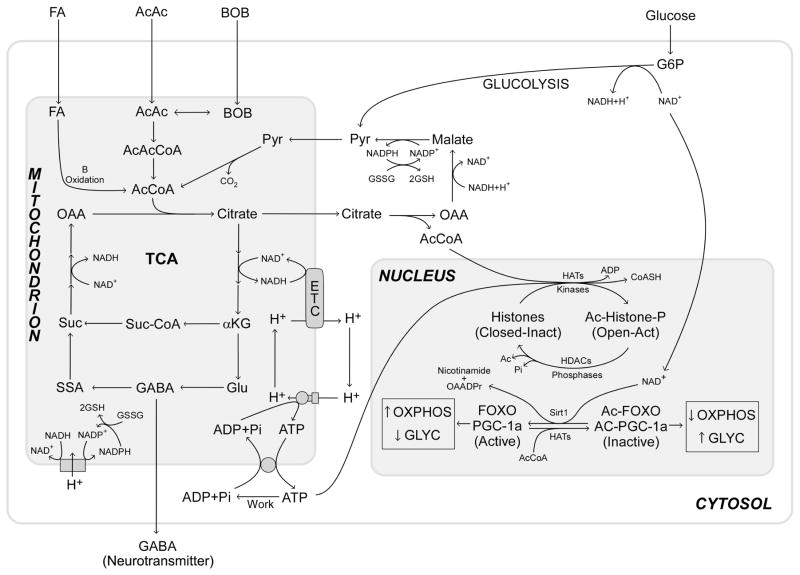
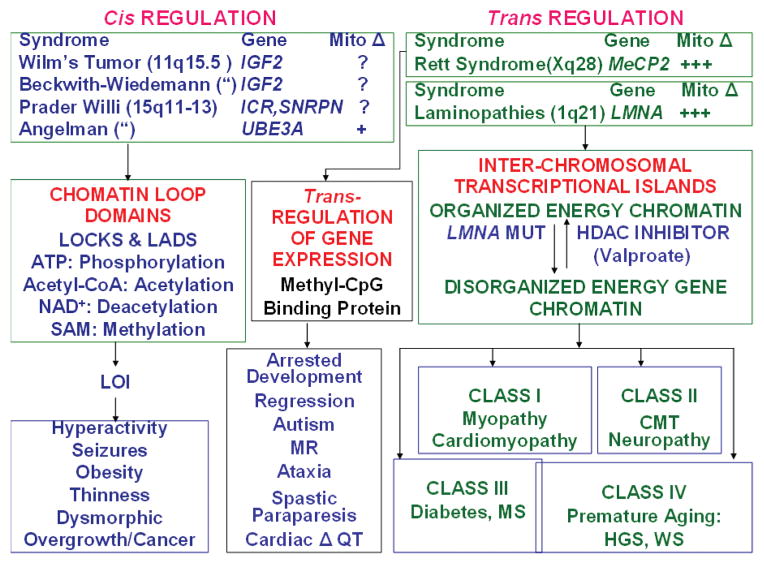
Studies on the origin of species have focused largely on anatomy, yet animal populations are generally limited by energy. Animals can adapt to available energy resources at three levels: (1) evolution of different anatomical forms between groups of animals through nuclear DNA (nDNA) mutations, permitting exploitation of alternative energy reservoirs and resulting in new species with novel niches, (2) evolution of different physiologies within intraspecific populations through mutations in mitochondrial DNA (mtDNA) and nDNA bioenergetic genes, permitting adjustment to energetic variation within a species' niche, and (3) epigenomic regulation of dispersed bioenergetic genes within an individual via mitochondrially generated high-energy intermediates, permitting individual adjustment to environmental fluctuations. Because medicine focuses on changes within our species, clinically relevant variation is more likely to involve changes in bioenergetics than anatomy. This may explain why mitochondrial diseases and epigenomic diseases frequently have similar phenotypes and why epigenomic diseases are being found to involve mitochondrial dysfunction. Therefore, common complex diseases may be the result of changes in any of a large number of mtDNA and nDNA bioenergetic genes or to altered epigenomic regulation of these bioenergetic genes. All of these changes result in similar bioenergetic failure and consequently related phenotypes.
Figures
Similar articles
-
Colloquium paper: bioenergetics, the origins of complexity, and the ascent of man.Proc Natl Acad Sci U S A. 2010 May 11;107 Suppl 2(Suppl 2):8947-53. doi: 10.1073/pnas.0914635107. Epub 2010 May 5. Proc Natl Acad Sci U S A. 2010. PMID: 20445102 Free PMC article.
-
Energetics, epigenetics, mitochondrial genetics.Mitochondrion. 2010 Jan;10(1):12-31. doi: 10.1016/j.mito.2009.09.006. Epub 2009 Sep 29. Mitochondrion. 2010. PMID: 19796712 Free PMC article. Review.
-
A mitochondrial bioenergetic etiology of disease.J Clin Invest. 2013 Apr;123(4):1405-12. doi: 10.1172/JCI61398. Epub 2013 Apr 1. J Clin Invest. 2013. PMID: 23543062 Free PMC article.
-
Bioenergetics and the epigenome: interface between the environment and genes in common diseases.Dev Disabil Res Rev. 2010;16(2):114-9. doi: 10.1002/ddrr.113. Dev Disabil Res Rev. 2010. PMID: 20818725 Review.
-
Mitochondrial DNA mutations in disease and aging.Environ Mol Mutagen. 2010 Jun;51(5):440-50. doi: 10.1002/em.20586. Environ Mol Mutagen. 2010. PMID: 20544884 Review.
Cited by
-
Circadian acetylome reveals regulation of mitochondrial metabolic pathways.Proc Natl Acad Sci U S A. 2013 Feb 26;110(9):3339-44. doi: 10.1073/pnas.1217632110. Epub 2013 Jan 22. Proc Natl Acad Sci U S A. 2013. PMID: 23341599 Free PMC article.
-
Quantitative proteomics of rat livers shows that unrestricted feeding is stressful for proteostasis with implications on life span.Aging (Albany NY). 2016 Aug;8(8):1735-58. doi: 10.18632/aging.101009. Aging (Albany NY). 2016. PMID: 27508340 Free PMC article.
-
The dynamic regulation of NAD metabolism in mitochondria.Trends Endocrinol Metab. 2012 Sep;23(9):420-8. doi: 10.1016/j.tem.2012.06.005. Epub 2012 Jul 21. Trends Endocrinol Metab. 2012. PMID: 22819213 Free PMC article.
-
Biological Aspects of Inflamm-Aging in Childhood Cancer Survivors.Cancers (Basel). 2021 Sep 30;13(19):4933. doi: 10.3390/cancers13194933. Cancers (Basel). 2021. PMID: 34638416 Free PMC article. Review.
-
Metabolic Plasticity in Cancer Cells: Reconnecting Mitochondrial Function to Cancer Control.J Cell Sci Ther. 2015 Jun;6(3):211. doi: 10.4172/2157-7013.1000211. Epub 2015 Jun 22. J Cell Sci Ther. 2015. PMID: 26457230 Free PMC article.
References
-
- Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, Lane CR, Schaffner SF, Bolk S, Brewer C, et al. The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000;26:76–80. - PubMed
-
- Cann RL, Stoneking M, Wilson AC. Mitochondrial DNA and human evolution. Nature. 1987;325:31–36. - PubMed
-
- Caron M, Auclair M, Donadille B, Bereziat V, Guerci B, Laville M, Narbonne H, Bodemer C, Lascols O, Capeau J, Vigouroux C. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007;14:1759–1767. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- NS21328/NS/NINDS NIH HHS/United States
- R01 AG024373-05/AG/NIA NIH HHS/United States
- AG16573/AG/NIA NIH HHS/United States
- R01 NS021328-24/NS/NINDS NIH HHS/United States
- R01 DK073691-04/DK/NIDDK NIH HHS/United States
- DK73691/DK/NIDDK NIH HHS/United States
- P50 AG016573/AG/NIA NIH HHS/United States
- R01 AG013154-11/AG/NIA NIH HHS/United States
- AG24373/AG/NIA NIH HHS/United States
- R01 AG024373/AG/NIA NIH HHS/United States
- R01 DK073691/DK/NIDDK NIH HHS/United States
- R01 NS021328/NS/NINDS NIH HHS/United States
- R01 AG013154/AG/NIA NIH HHS/United States
- AG13154/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources