

Embryonic lethality after combined inactivation of Fancd2 and Mlh1 in mice
- PMID: 19934329
- PMCID: PMC2799030
- DOI: 10.1158/0008-5472.CAN-09-2452
Embryonic lethality after combined inactivation of Fancd2 and Mlh1 in mice
Abstract
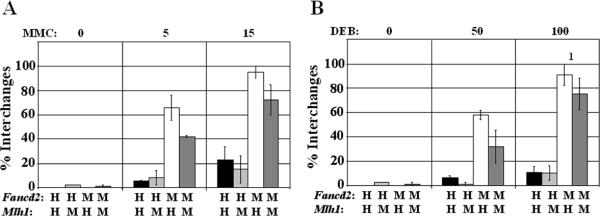
DNA repair defects are frequently encountered in human cancers. These defects are utilized by traditional therapeutics but also offer novel cancer treatment strategies based on synthetic lethality. To determine the consequences of combined Fanconi anemia (FA) and mismatch repair pathway inactivation, defects in Fancd2 and Mlh1 were combined in one mouse model. Fancd2/Mlh1 double-mutant embryos displayed growth retardation resulting in embryonic lethality and significant underrepresentation among progeny. Additional inactivation of Trp53 failed to improve the survival of Fancd2/Mlh1-deficient embryos. Mouse fibroblasts were obtained and challenged with cross-linking agents. Fancd2-deficient cells displayed the FA-characteristic growth inhibition after mitomycin C (MMC) exposure. In primary fibroblasts, the absence of Mlh1 did not greatly affect the MMC sensitivity of Fancd2-deficient and Fancd2-proficient cells. However, in Trp53 mutant immortalized fibroblasts, Mlh1 deficiency reduced the growth-inhibiting effect of MMC in Fancd2 mutant and complemented cells. Similar data were obtained using psoralen/UVA, signifying that MLH1 influences the cellular sensitivity to DNA interstrand cross-links. Next, the effect of MLH1 deficiency on the formation of chromosomal aberrations in response to cross-linking agents was determined. Surprisingly, Mlh1 mutant fibroblasts displayed a modest but noticeable decrease in induced chromosomal breakage and interchange frequencies, suggesting that MLH1 promotes interstrand cross-link repair catastrophe. In conclusion, the combined inactivation of Fancd2 and Mlh1 did not result in synthetic lethality at the cellular level. Although the absence of Fancd2 sensitized Mlh1/Trp53 mutant fibroblasts to MMC, the differential survival of primary and immortalized fibroblasts advocates against systemic inactivation of FANCD2 to enhance treatment of MLH1-deficient tumors.
Figures
Similar articles
-
Crosstalk between BRCA-Fanconi anemia and mismatch repair pathways prevents MSH2-dependent aberrant DNA damage responses.EMBO J. 2014 Aug 1;33(15):1698-712. doi: 10.15252/embj.201387530. Epub 2014 Jun 25. EMBO J. 2014. PMID: 24966277 Free PMC article.
-
Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice.Genes Dev. 2003 Aug 15;17(16):2021-35. doi: 10.1101/gad.1103403. Epub 2003 Jul 31. Genes Dev. 2003. PMID: 12893777 Free PMC article.
-
Heterozygosity for p53 (Trp53+/-) accelerates epithelial tumor formation in fanconi anemia complementation group D2 (Fancd2) knockout mice.Cancer Res. 2005 Jan 1;65(1):85-91. Cancer Res. 2005. PMID: 15665282
-
The Fanconi anemia ID2 complex: dueling saxes at the crossroads.Cell Cycle. 2014;13(19):2999-3015. doi: 10.4161/15384101.2014.956475. Cell Cycle. 2014. PMID: 25486561 Free PMC article. Review.
-
Current knowledge on the pathophysiology of Fanconi anemia: from genes to phenotypes.Int J Hematol. 2001 Jul;74(1):33-41. doi: 10.1007/BF02982547. Int J Hematol. 2001. PMID: 11530803 Review.
Cited by
-
Fanconi anemia signaling and Mus81 cooperate to safeguard development and crosslink repair.Nucleic Acids Res. 2014 Sep;42(15):9807-20. doi: 10.1093/nar/gku676. Epub 2014 Jul 23. Nucleic Acids Res. 2014. PMID: 25056314 Free PMC article.
-
Crosstalk between BRCA-Fanconi anemia and mismatch repair pathways prevents MSH2-dependent aberrant DNA damage responses.EMBO J. 2014 Aug 1;33(15):1698-712. doi: 10.15252/embj.201387530. Epub 2014 Jun 25. EMBO J. 2014. PMID: 24966277 Free PMC article.
-
Learning from a paradox: recent insights into Fanconi anaemia through studying mouse models.Dis Model Mech. 2013 Jan;6(1):40-7. doi: 10.1242/dmm.009795. Dis Model Mech. 2013. PMID: 23268537 Free PMC article. Review.
References
-
- Hoeijmakers JHJ. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–74. - PubMed
-
- Van de Vrugt HJ, Grompe M. Fanconi anemia. In: Epstein CJ, Erickson RP, Wynshaw-Boris A, editors. Inborn errors of development. 2nd ed. Oxford University Press; New York: 2008. pp. 1230–6.
-
- Chirnomas D, Taniguchi T, De la Vega M, Vaidya AP, Vasserman M, Hartman AR, et al. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol Cancer Ther. 2006;5:952–61. - PubMed
-
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. - PubMed
-
- McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–15. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
