

Molecular basis for the recognition and cleavages of IGF-II, TGF-alpha, and amylin by human insulin-degrading enzyme
- PMID: 19896952
- PMCID: PMC2813390
- DOI: 10.1016/j.jmb.2009.10.072
Molecular basis for the recognition and cleavages of IGF-II, TGF-alpha, and amylin by human insulin-degrading enzyme
Abstract
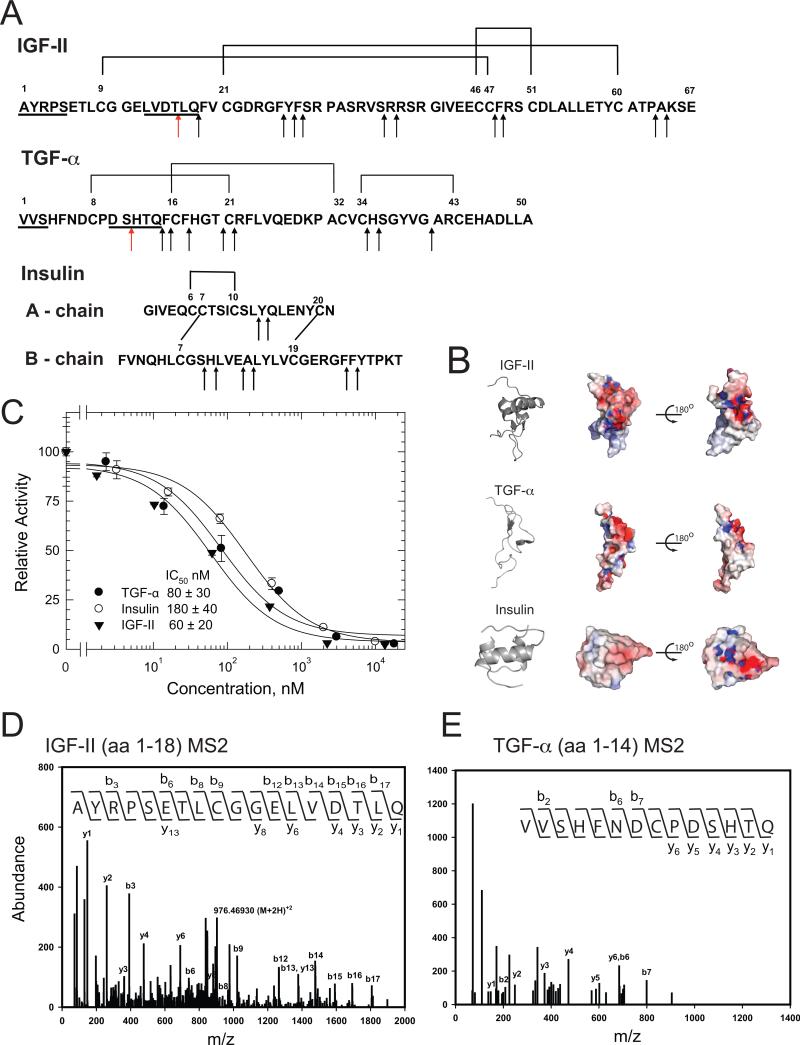
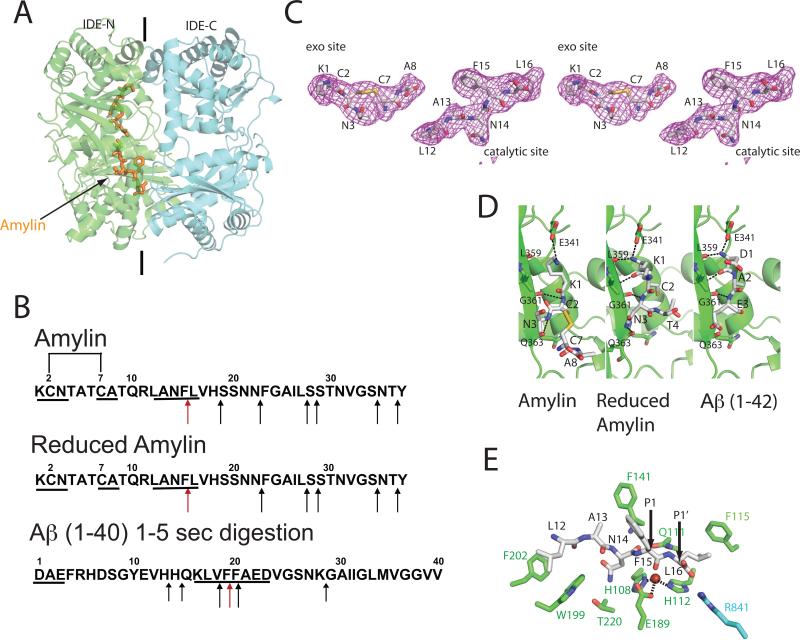
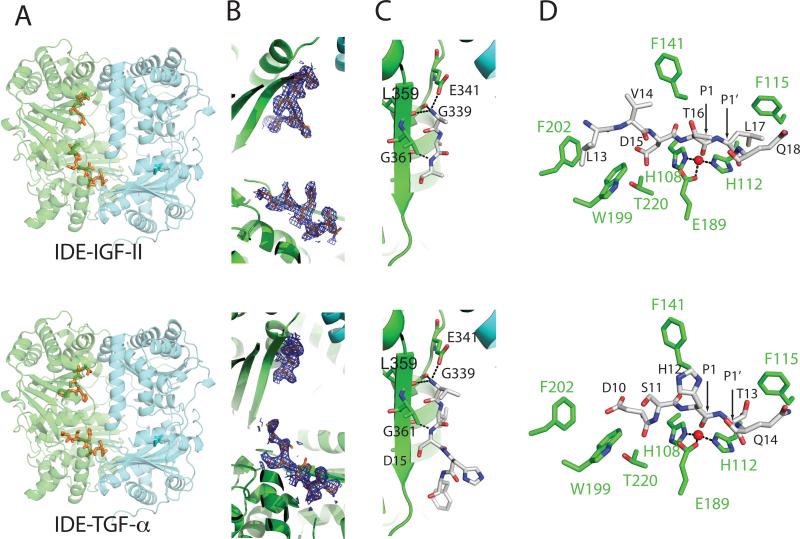
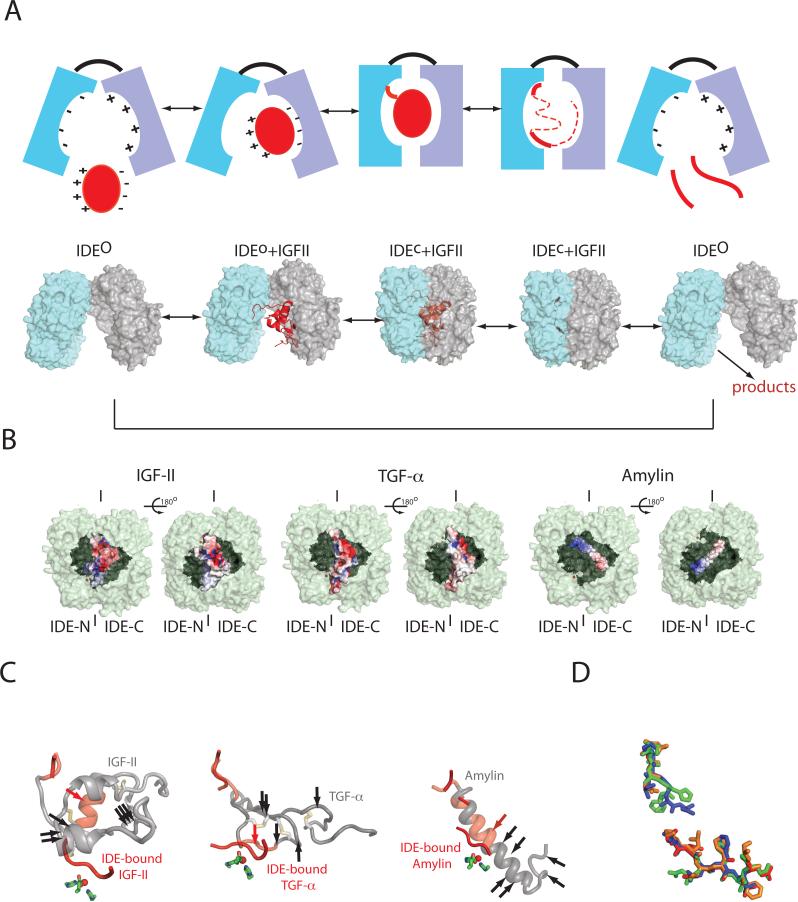
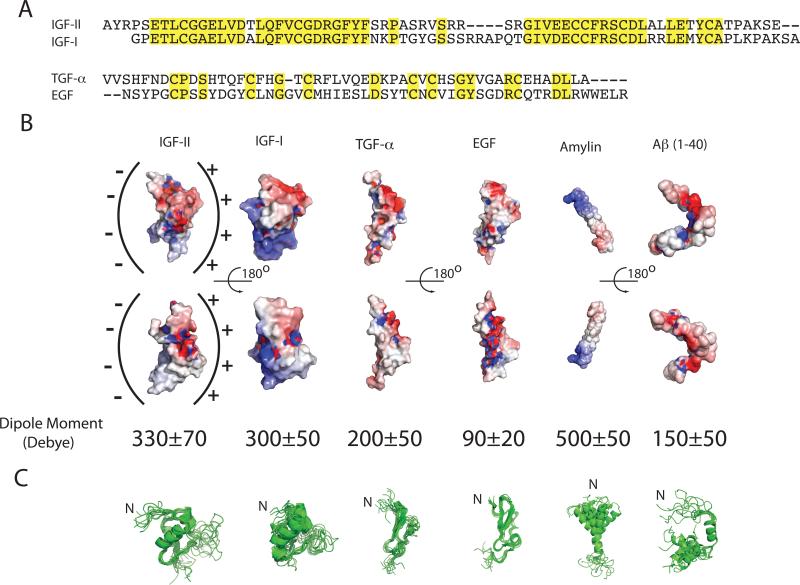
Insulin-degrading enzyme (IDE) is involved in the clearance of many bioactive peptide substrates, including insulin and amyloid-beta, peptides vital to the development of diabetes and Alzheimer's disease, respectively. IDE can also rapidly degrade hormones that are held together by intramolecular disulfide bond(s) without their reduction. Furthermore, IDE exhibits a remarkable ability to preferentially degrade structurally similar peptides such as the selective degradation of insulin-like growth factor (IGF)-II and transforming growth factor-alpha (TGF-alpha) over IGF-I and epidermal growth factor, respectively. Here, we used high-accuracy mass spectrometry to identify the cleavage sites of human IGF-II, TGF-alpha, amylin, reduced amylin, and amyloid-beta by human IDE. We also determined the structures of human IDE-IGF-II and IDE-TGF-alpha at 2.3 A and IDE-amylin at 2.9 A. We found that IDE cleaves its substrates at multiple sites in a biased stochastic manner. Furthermore, the presence of a disulfide bond in amylin allows IDE to cut at an additional site in the middle of the peptide (amino acids 18-19). Our amylin-bound IDE structure offers insight into how the structural constraint from a disulfide bond in amylin can alter IDE cleavage sites. Together with NMR structures of amylin and the IGF and epidermal growth factor families, our work also reveals the structural basis of how the high dipole moment of substrates complements the charge distribution of the IDE catalytic chamber for the substrate selectivity. In addition, we show how the ability of substrates to properly anchor their N-terminus to the exosite of IDE and undergo a conformational switch upon binding to the catalytic chamber of IDE can also contribute to the selective degradation of structurally related growth factors.
Copyright 2009 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism.Nature. 2006 Oct 19;443(7113):870-4. doi: 10.1038/nature05143. Epub 2006 Oct 11. Nature. 2006. PMID: 17051221 Free PMC article.
-
Molecular basis of catalytic chamber-assisted unfolding and cleavage of human insulin by human insulin-degrading enzyme.J Biol Chem. 2009 May 22;284(21):14177-88. doi: 10.1074/jbc.M900068200. Epub 2009 Mar 25. J Biol Chem. 2009. PMID: 19321446 Free PMC article.
-
Molecular bases for the recognition of short peptide substrates and cysteine-directed modifications of human insulin-degrading enzyme.Biochemistry. 2008 Dec 2;47(48):12822-34. doi: 10.1021/bi801192h. Biochemistry. 2008. PMID: 18986166 Free PMC article.
-
Insulin-degrading enzyme: structure-function relationship and its possible roles in health and disease.Curr Pharm Des. 2009;15(31):3644-55. doi: 10.2174/138161209789271799. Curr Pharm Des. 2009. PMID: 19925417 Review.
-
Targeting Insulin-Degrading Enzyme to Treat Type 2 Diabetes Mellitus.Trends Endocrinol Metab. 2016 Jan;27(1):24-34. doi: 10.1016/j.tem.2015.11.003. Epub 2015 Dec 2. Trends Endocrinol Metab. 2016. PMID: 26651592 Free PMC article. Review.
Cited by
-
Linking hIAPP misfolding and aggregation with type 2 diabetes mellitus: a structural perspective.Biosci Rep. 2022 May 27;42(5):BSR20211297. doi: 10.1042/BSR20211297. Biosci Rep. 2022. PMID: 35475576 Free PMC article. Review.
-
Quantitative NMR Study of Insulin-Degrading Enzyme Using Amyloid-β and HIV-1 p6 Elucidates Its Chaperone Activity.Biochemistry. 2021 Aug 24;60(33):2519-2523. doi: 10.1021/acs.biochem.1c00342. Epub 2021 Aug 3. Biochemistry. 2021. PMID: 34342986 Free PMC article.
-
Identification of disulfide bonds in protein proteolytic degradation products using de novo-protein unique sequence tags approach.J Proteome Res. 2010 Aug 6;9(8):4053-60. doi: 10.1021/pr1002559. J Proteome Res. 2010. PMID: 20590115 Free PMC article.
-
An Extended Polyanion Activation Surface in Insulin Degrading Enzyme.PLoS One. 2015 Jul 17;10(7):e0133114. doi: 10.1371/journal.pone.0133114. eCollection 2015. PLoS One. 2015. PMID: 26186535 Free PMC article.
-
Characterization of a Species-Specific Insulinase-Like Protease in Cryptosporidium parvum.Front Microbiol. 2019 Mar 6;10:354. doi: 10.3389/fmicb.2019.00354. eCollection 2019. Front Microbiol. 2019. PMID: 30894838 Free PMC article.
References
-
- Duckworth WC, Bennett RG, Hamel FG. Insulin degradation: progress and potential. Endocr Rev. 1998;19:608–24. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
