

Spinocerebellar ataxia type 31 is associated with "inserted" penta-nucleotide repeats containing (TGGAA)n
- PMID: 19878914
- PMCID: PMC2775824
- DOI: 10.1016/j.ajhg.2009.09.019
Spinocerebellar ataxia type 31 is associated with "inserted" penta-nucleotide repeats containing (TGGAA)n
Abstract
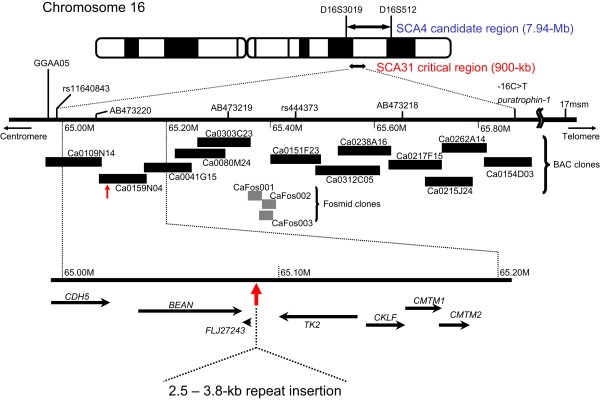
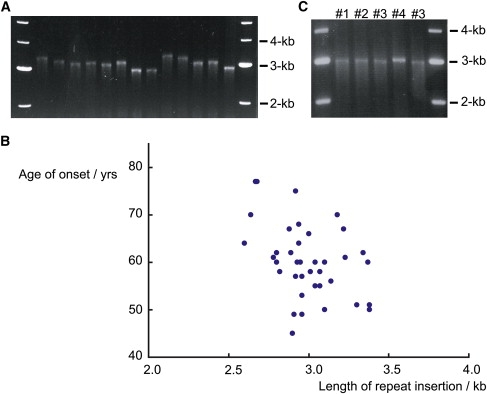
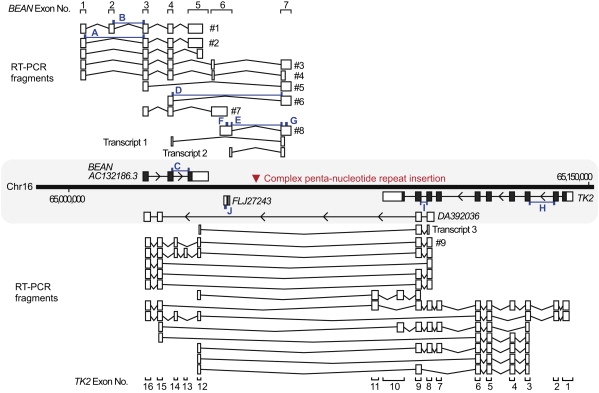
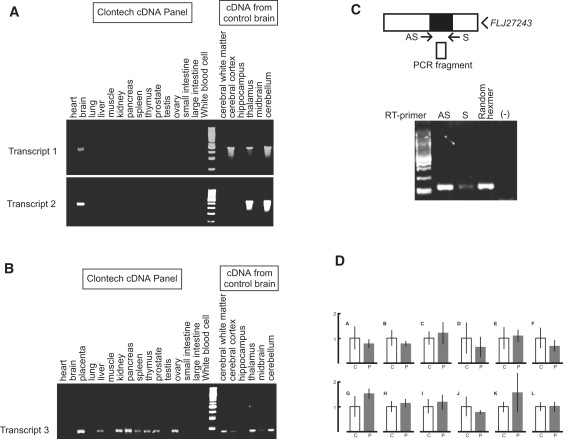
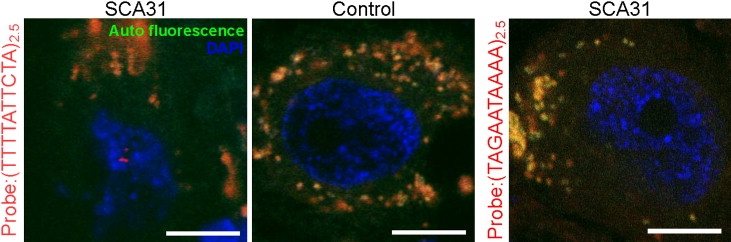
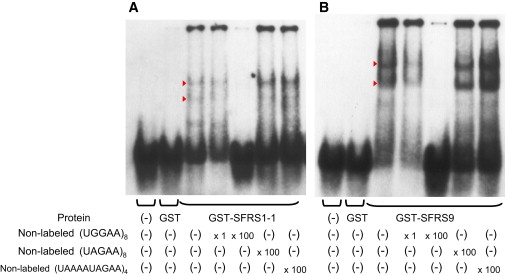
Spinocerebellar ataxia type 31 (SCA31) is an adult-onset autosomal-dominant neurodegenerative disorder showing progressive cerebellar ataxia mainly affecting Purkinje cells. The SCA31 critical region was tracked down to a 900 kb interval in chromosome 16q22.1, where the disease shows a strong founder effect. By performing comprehensive Southern blot analysis and BAC- and fosmid-based sequencing, we isolated two genetic changes segregating with SCA31. One was a single-nucleotide change in an intron of the thymidine kinase 2 gene (TK2). However, this did not appear to affect splicing or expression patterns. The other was an insertion, from 2.5-3.8 kb long, consisting of complex penta-nucleotide repeats including a long (TGGAA)n stretch. In controls, shorter (1.5-2.0 kb) insertions lacking (TGGAA)n were found only rarely. The SCA31 repeat insertion's length inversely correlated with patient age of onset, and an expansion was documented in a single family showing anticipation. The repeat insertion was located in introns of TK2 and BEAN (brain expressed, associated with Nedd4) expressed in the brain and formed RNA foci in the nuclei of patients' Purkinje cells. An electrophoretic mobility-shift assay showed that essential splicing factors, serine/arginine-rich splicing factors SFRS1 and SFRS9, bind to (UGGAA)n in vitro. Because (TGGAA)n is a characteristic sequence of paracentromeric heterochromatin, we speculate that the insertion might have originated from heterochromatin. SCA31 is important because it exemplifies human diseases associated with "inserted" microsatellite repeats that can expand through transmission. Our finding suggests that the ectopic microsatellite repeat, when transcribed, might cause a disease involving the essential splicing factors.
Figures

Similar articles
-
[Spinocerebellar ataxia type 31].Rinsho Shinkeigaku. 2010 Nov;50(11):985-7. doi: 10.5692/clinicalneurol.50.985. Rinsho Shinkeigaku. 2010. PMID: 21921537 Review. Japanese.
-
Abnormal RNA structures (RNA foci) containing a penta-nucleotide repeat (UGGAA)n in the Purkinje cell nucleus is associated with spinocerebellar ataxia type 31 pathogenesis.Neuropathology. 2013 Dec;33(6):600-11. doi: 10.1111/neup.12032. Epub 2013 Apr 22. Neuropathology. 2013. PMID: 23607545
-
Molecular Mechanisms and Future Therapeutics for Spinocerebellar Ataxia Type 31 (SCA31).Neurotherapeutics. 2019 Oct;16(4):1106-1114. doi: 10.1007/s13311-019-00804-6. Neurotherapeutics. 2019. PMID: 31755042 Free PMC article. Review.
-
Analysis of an insertion mutation in a cohort of 94 patients with spinocerebellar ataxia type 31 from Nagano, Japan.Neurogenetics. 2010 Oct;11(4):409-15. doi: 10.1007/s10048-010-0245-6. Epub 2010 Apr 28. Neurogenetics. 2010. PMID: 20424877 Free PMC article.
-
Spinocerebellar ataxia type 31 (SCA31).J Hum Genet. 2023 Mar;68(3):153-156. doi: 10.1038/s10038-022-01091-4. Epub 2022 Nov 1. J Hum Genet. 2023. PMID: 36319738 Free PMC article. Review.
Cited by
-
RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy.Mol Ther. 2013 Feb;21(2):380-7. doi: 10.1038/mt.2012.222. Epub 2012 Nov 27. Mol Ther. 2013. PMID: 23183533 Free PMC article.
-
Distinct roles for Toll and autophagy pathways in double-stranded RNA toxicity in a Drosophila model of expanded repeat neurodegenerative diseases.Hum Mol Genet. 2013 Jul 15;22(14):2811-9. doi: 10.1093/hmg/ddt130. Epub 2013 Mar 21. Hum Mol Genet. 2013. PMID: 23525903 Free PMC article.
-
Autosomal dominant cerebellar ataxia type III: a review of the phenotypic and genotypic characteristics.Orphanet J Rare Dis. 2013 Jan 18;8:14. doi: 10.1186/1750-1172-8-14. Orphanet J Rare Dis. 2013. PMID: 23331413 Free PMC article. Review.
-
Long-read sequencing for rare human genetic diseases.J Hum Genet. 2020 Jan;65(1):11-19. doi: 10.1038/s10038-019-0671-8. Epub 2019 Sep 27. J Hum Genet. 2020. PMID: 31558760 Review.
-
Two Italian families with ITPR1 gene deletion presenting a broader phenotype of SCA15.Cerebellum. 2010 Mar;9(1):115-23. doi: 10.1007/s12311-009-0154-0. Cerebellum. 2010. PMID: 20082166
References
-
- Brice A., Pulst S.-M. Butterworth Heinemann, Elsevier, Inc.; Philadelphia: 2007. Spinocerebellar degenerations: The ataxias and spastic paraplegias.
-
- Zoghbi H.Y., Orr H.T. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000;23:217–247. - PubMed
-
- Ranum L.P.W., Cooper T.A. RNA-mediated neuromuscular disorders. Annu. Rev. Neurosci. 2006;29:259–277. - PubMed
-
- Bandmann O., Singleton A.B. Yet another spinocerebellar ataxia: The saga continues. Neurology. 2008;71:542–543. - PubMed
-
- Nagaoka U., Takashima M., Ishikawa K., Yoshizawa K., Yoshizawa T., Ishikawa M., Yamawaki T., Shoji S., Mizusawa H. A gene on SCA4 locus causes dominantly inherited pure cerebellar ataxia. Neurology. 2000;54:1971–1975. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
