

Mechanisms of the epithelial-mesenchymal transition by TGF-beta
- PMID: 19852727
- PMCID: PMC2858056
- DOI: 10.2217/fon.09.90
Mechanisms of the epithelial-mesenchymal transition by TGF-beta
Abstract
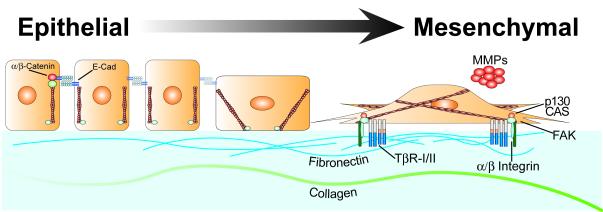
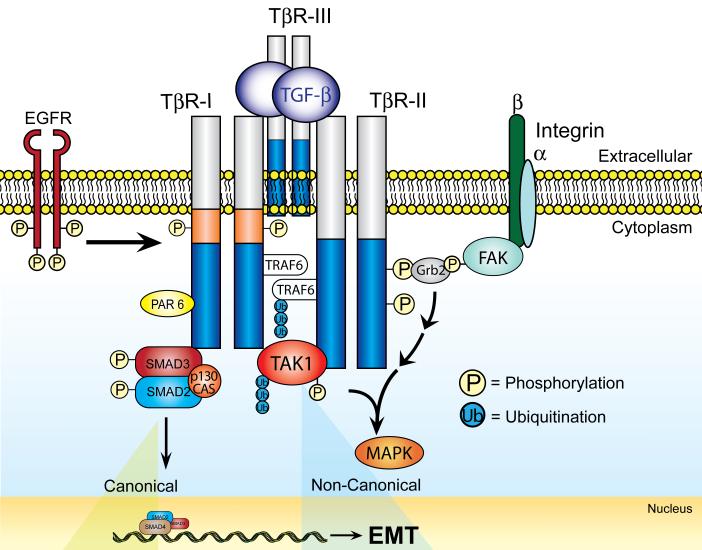
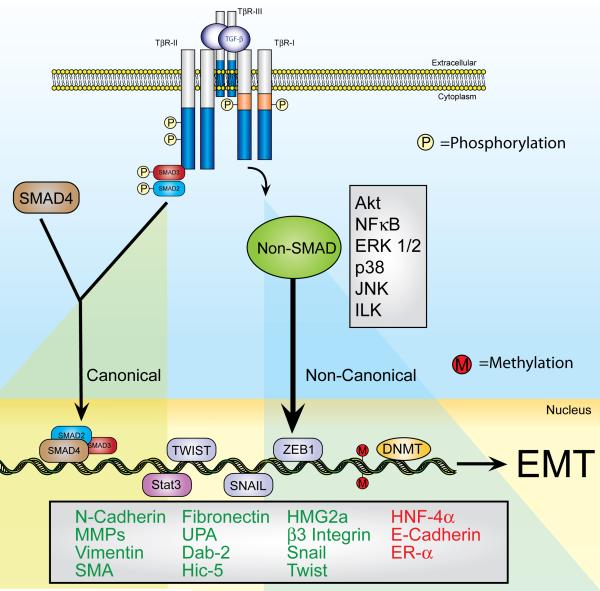
The formation of epithelial cell barriers results from the defined spatiotemporal differentiation of stem cells into a specialized and polarized epithelium, a process termed mesenchymal-epithelial transition. The reverse process, epithelial-mesenchymal transition (EMT), is a metastable process that enables polarized epithelial cells to acquire a motile fibroblastoid phenotype. Physiological EMT also plays an essential role in promoting tissue healing, remodeling or repair in response to a variety of pathological insults. On the other hand, pathophysiological EMT is a critical step in mediating the acquisition of metastatic phenotypes by localized carcinomas. Although metastasis clearly is the most lethal aspect of cancer, our knowledge of the molecular events that govern its development, including those underlying EMT, remain relatively undefined. Transforming growth factor-beta (TGF-beta) is a multifunctional cytokine that oversees and directs all aspects of cell development, differentiation and homeostasis, as well as suppresses their uncontrolled proliferation and transformation. Quite dichotomously, tumorigenesis subverts the tumor suppressing function of TGF-beta, and in doing so, converts TGF-beta to a tumor promoter that stimulates pathophysiological EMT and metastasis. It therefore stands to reason that determining how TGF-beta induces EMT in developing neoplasms will enable science and medicine to produce novel pharmacological agents capable of preventing its ability to do so, thereby improving the clinical course of cancer patients. Here we review the cellular, molecular and microenvironmental mechanisms used by TGF-beta to mediate its stimulation of EMT in normal and malignant cells.
Figures
Similar articles
-
The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells.J Mammary Gland Biol Neoplasia. 2010 Jun;15(2):169-90. doi: 10.1007/s10911-010-9181-1. Epub 2010 May 15. J Mammary Gland Biol Neoplasia. 2010. PMID: 20467795 Free PMC article. Review.
-
Epithelial-mesenchymal transition in hepatocellular carcinoma.Future Oncol. 2009 Oct;5(8):1169-79. doi: 10.2217/fon.09.91. Future Oncol. 2009. PMID: 19852728 Free PMC article. Review.
-
Epithelial-mesenchymal transition in development and cancer.Future Oncol. 2009 Oct;5(8):1129-43. doi: 10.2217/fon.09.94. Future Oncol. 2009. PMID: 19852726 Review.
-
Fibulin-5 initiates epithelial-mesenchymal transition (EMT) and enhances EMT induced by TGF-beta in mammary epithelial cells via a MMP-dependent mechanism.Carcinogenesis. 2008 Dec;29(12):2243-51. doi: 10.1093/carcin/bgn199. Epub 2008 Aug 19. Carcinogenesis. 2008. PMID: 18713838 Free PMC article.
-
Na,K-ATPase subunits as markers for epithelial-mesenchymal transition in cancer and fibrosis.Mol Cancer Ther. 2010 Jun;9(6):1515-24. doi: 10.1158/1535-7163.MCT-09-0832. Epub 2010 May 25. Mol Cancer Ther. 2010. PMID: 20501797 Free PMC article.
Cited by
-
Integrative genome-wide gene expression profiling of clear cell renal cell carcinoma in Czech Republic and in the United States.PLoS One. 2013;8(3):e57886. doi: 10.1371/journal.pone.0057886. Epub 2013 Mar 5. PLoS One. 2013. PMID: 23526956 Free PMC article.
-
Transcriptome Landscape of Epithelial to Mesenchymal Transition of Human Stem Cell-Derived RPE.Invest Ophthalmol Vis Sci. 2021 Apr 1;62(4):1. doi: 10.1167/iovs.62.4.1. Invest Ophthalmol Vis Sci. 2021. PMID: 33792620 Free PMC article.
-
Inhibition of TGF-β signaling enables human corneal endothelial cell expansion in vitro for use in regenerative medicine.PLoS One. 2013;8(2):e58000. doi: 10.1371/journal.pone.0058000. Epub 2013 Feb 25. PLoS One. 2013. PMID: 23451286 Free PMC article.
-
Phenotypic Transition as a Survival Strategy of Glioma.Neurol Med Chir (Tokyo). 2016 Jul 15;56(7):387-95. doi: 10.2176/nmc.ra.2016-0077. Epub 2016 May 11. Neurol Med Chir (Tokyo). 2016. PMID: 27169497 Free PMC article. Review.
-
Chemotherapeutic Targeting of the Transforming Growth Factor-β Pathway in Breast Cancers.Breast Cancer Manag. 2014;3(1):73-85. doi: 10.2217/bmt.13.74. Breast Cancer Manag. 2014. PMID: 25904986 Free PMC article.
References
-
- Baum B, Settleman J, Quinlan MP. Transitions between epithelial and mesenchymal states in development and disease. Semin. Cell Dev. Biol. 2008;19:294–308. - PubMed
-
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. Cell. 2008;14:818–829. - PubMed
-
- Elenbaas B, Weinberg RA. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp. Cell Res. 2001;264:169–184. - PubMed
-
- Zavadil J, Bottinger EP. TGF-β and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources