

Effects of Src inhibitors on cell growth and epidermal growth factor receptor and MET signaling in gefitinib-resistant non-small cell lung cancer cells with acquired MET amplification
- PMID: 19804422
- PMCID: PMC11158912
- DOI: 10.1111/j.1349-7006.2009.01368.x
Effects of Src inhibitors on cell growth and epidermal growth factor receptor and MET signaling in gefitinib-resistant non-small cell lung cancer cells with acquired MET amplification
Abstract
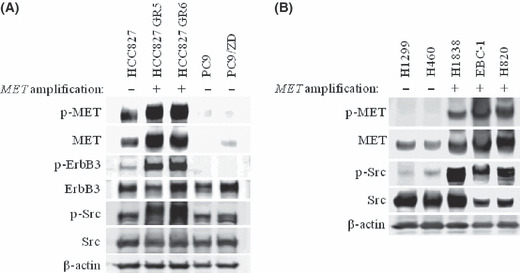
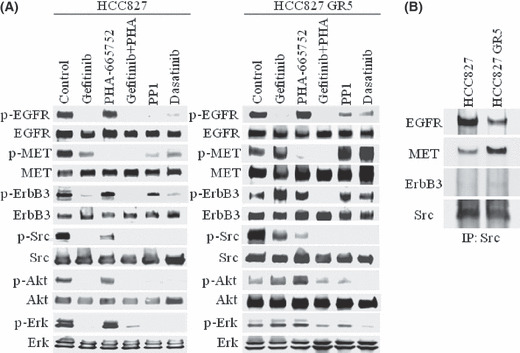
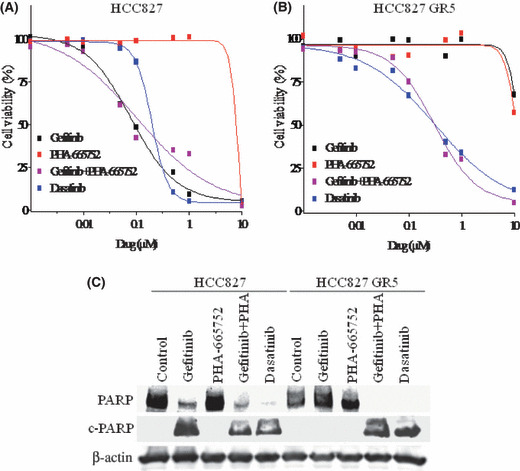
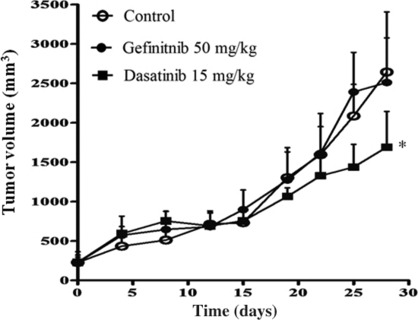
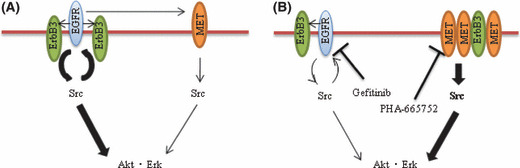
The efficacy of epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitors such as gefitinib and erlotinib in non-small cell lung cancer (NSCLC) is often limited by the emergence of drug resistance conferred either by a secondary T790M mutation of EGFR or by acquired amplification of the MET gene. We now show that the extent of activation of the tyrosine kinase Src is markedly increased in gefitinib-resistant NSCLC (HCC827 GR) cells with MET amplification compared with that in the gefitinib-sensitive parental (HCC827) cells. In contrast, the extent of Src activation did not differ between gefitinib-resistant NSCLC (PC9/ZD) cells harboring the T790M mutation of EGFR and the corresponding gefitinib-sensitive parental (PC9) cells. This activation of Src in HCC827 GR cells was largely abolished by the MET-TKI PHA-665752 but was only partially inhibited by gefitinib, suggesting that Src activation is more dependent on MET signaling than on EGFR signaling in gefitinib-resistant NSCLC cells with MET amplification. Src inhibitors blocked Akt and Erk signaling pathways, resulting in both suppression of cell growth and induction of apoptosis, in HCC827 GR cells as effectively as did the combination of gefitinib and PHA-665752. Furthermore, Src inhibitor dasatinib inhibited tumor growth in HCC827 GR xenografts to a significantly greater extent than did treatment with gefitinib alone. These results provide a rationale for clinical targeting of Src in gefitinib-resistant NSCLC with MET amplification.
Figures
Similar articles
-
MET and AXL inhibitor NPS-1034 exerts efficacy against lung cancer cells resistant to EGFR kinase inhibitors because of MET or AXL activation.Cancer Res. 2014 Jan 1;74(1):253-62. doi: 10.1158/0008-5472.CAN-13-1103. Epub 2013 Oct 28. Cancer Res. 2014. PMID: 24165158
-
Reciprocal and complementary role of MET amplification and EGFR T790M mutation in acquired resistance to kinase inhibitors in lung cancer.Clin Cancer Res. 2010 Nov 15;16(22):5489-98. doi: 10.1158/1078-0432.CCR-10-1371. Epub 2010 Nov 9. Clin Cancer Res. 2010. PMID: 21062933
-
MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling.Science. 2007 May 18;316(5827):1039-43. doi: 10.1126/science.1141478. Epub 2007 Apr 26. Science. 2007. PMID: 17463250
-
Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway.Clin Lung Cancer. 2009 Jul;10(4):281-9. doi: 10.3816/CLC.2009.n.039. Clin Lung Cancer. 2009. PMID: 19632948 Free PMC article. Review.
-
Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors.Oncogene. 2009 Aug;28 Suppl 1(Suppl 1):S24-31. doi: 10.1038/onc.2009.198. Oncogene. 2009. PMID: 19680293 Free PMC article. Review.
Cited by
-
Single-cell lipidomics with high structural specificity by mass spectrometry.Nat Commun. 2021 May 17;12(1):2869. doi: 10.1038/s41467-021-23161-5. Nat Commun. 2021. PMID: 34001877 Free PMC article.
-
SRC and PIM1 as potential co-targets to overcome resistance in MET deregulated non-small cell lung cancer.Transl Lung Cancer Res. 2020 Oct;9(5):1810-1821. doi: 10.21037/tlcr-20-681. Transl Lung Cancer Res. 2020. PMID: 33209603 Free PMC article.
-
Combined treatment with N-acetylcysteine and gefitinib overcomes drug resistance to gefitinib in NSCLC cell line.Cancer Med. 2020 Feb;9(4):1495-1502. doi: 10.1002/cam4.2610. Epub 2019 Dec 31. Cancer Med. 2020. PMID: 31891230 Free PMC article.
-
Characterization of In Vivo Resistance to Osimertinib and JNJ-61186372, an EGFR/Met Bispecific Antibody, Reveals Unique and Consensus Mechanisms of Resistance.Mol Cancer Ther. 2017 Nov;16(11):2572-2585. doi: 10.1158/1535-7163.MCT-17-0413. Epub 2017 Aug 22. Mol Cancer Ther. 2017. PMID: 28830985 Free PMC article.
-
Sorting nexin 2-mediated membrane trafficking of c-Met contributes to sensitivity of molecular-targeted drugs.Cancer Sci. 2013 May;104(5):573-83. doi: 10.1111/cas.12117. Epub 2013 Mar 8. Cancer Sci. 2013. PMID: 23360489 Free PMC article.
References
-
- Gullick WJ. Prevalence of aberrant expression of the epidermal growth factor receptor in human cancers. Br Med Bull 1991; 47: 87–98. - PubMed
-
- Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor‐related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 1995; 19: 183–232. - PubMed
-
- Harari PM. Epidermal growth factor receptor inhibition strategies in oncology. Endocr Relat Cancer 2004; 11: 689–708. - PubMed
-
- Ettinger DS. Clinical implications of EGFR expression in the development and progression of solid tumors: focus on non‐small cell lung cancer. Oncologist 2006; 11: 358–73. - PubMed
-
- Lynch TJ, Bell DW, Sordella R et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
