

Extracellular superoxide dismutase regulates cardiac function and fibrosis
- PMID: 19695260
- PMCID: PMC2774793
- DOI: 10.1016/j.yjmcc.2009.08.010
Extracellular superoxide dismutase regulates cardiac function and fibrosis
Abstract
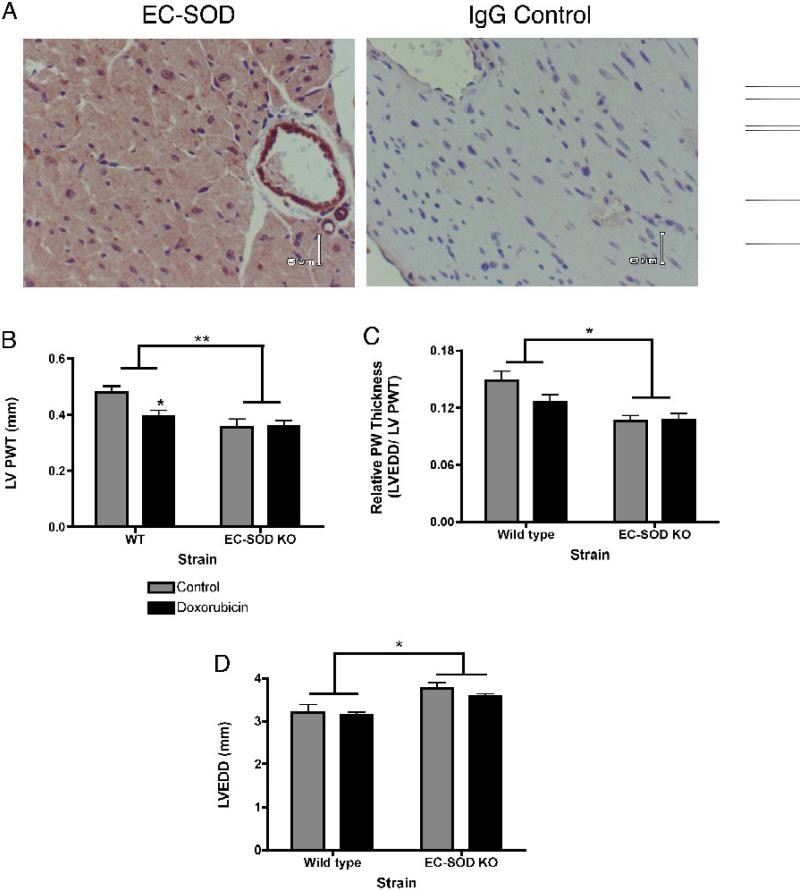
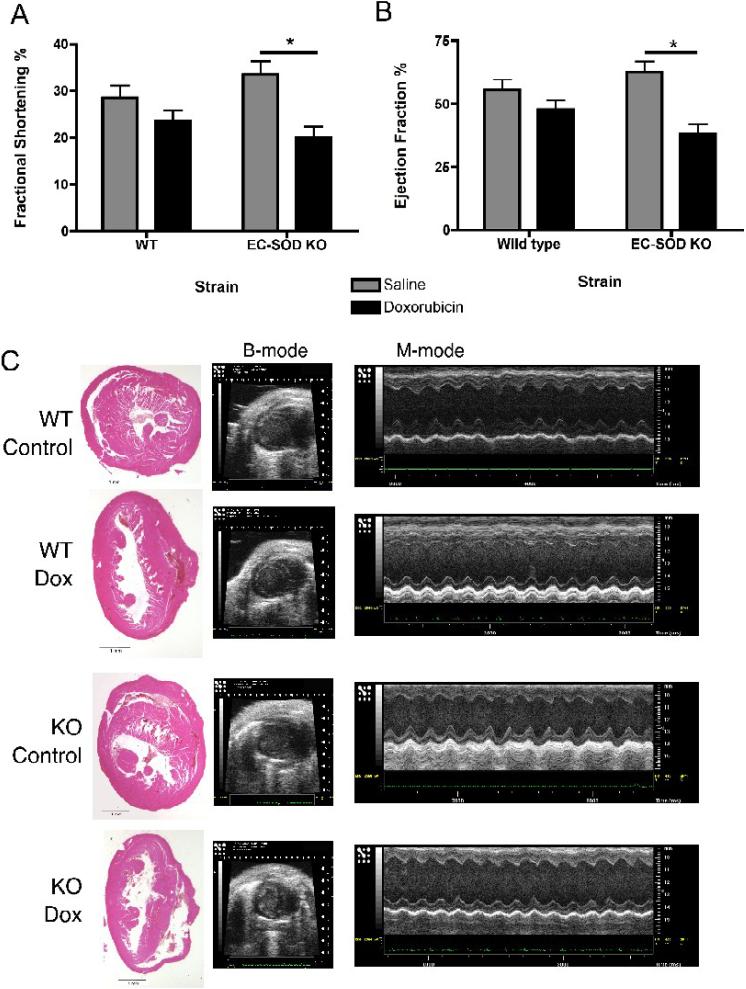
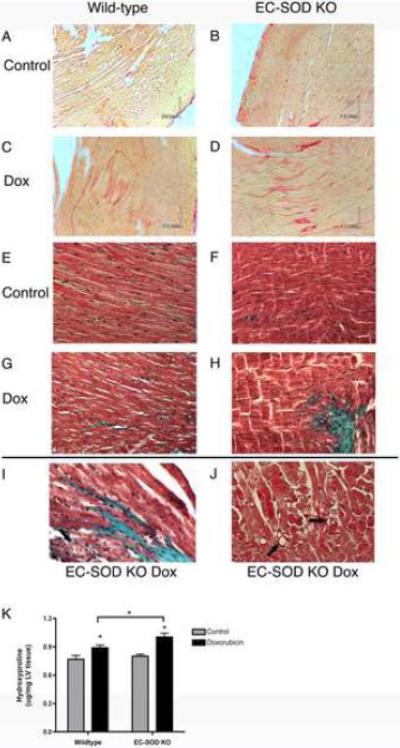
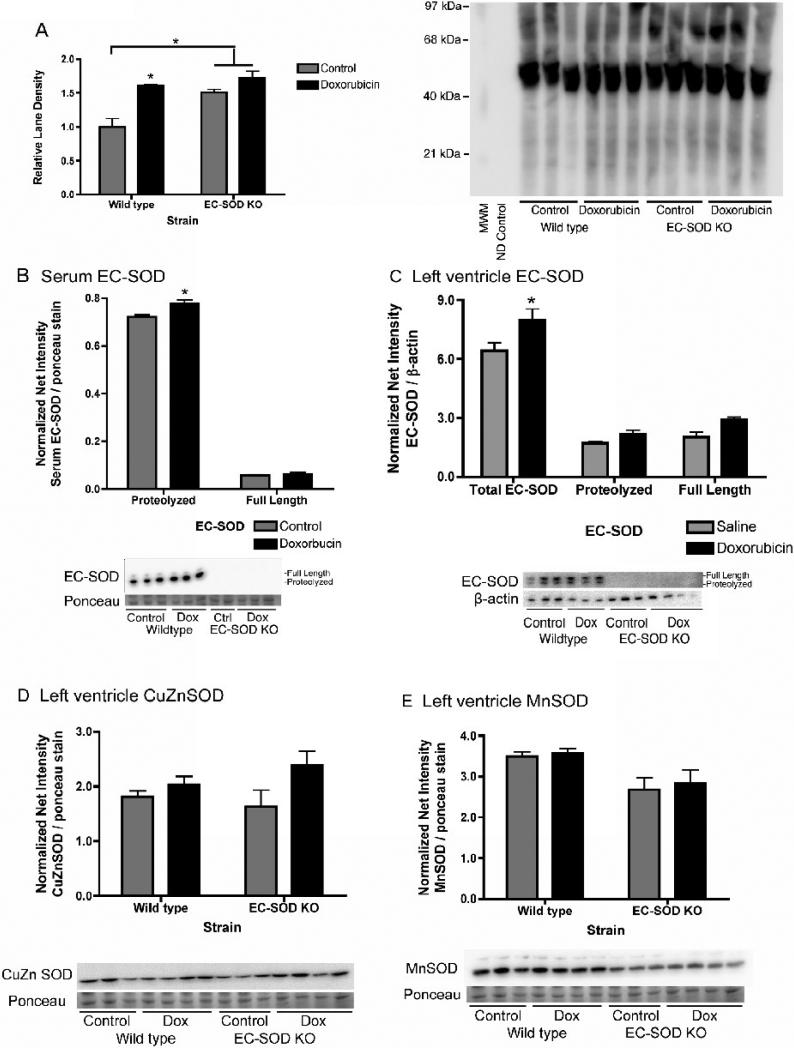
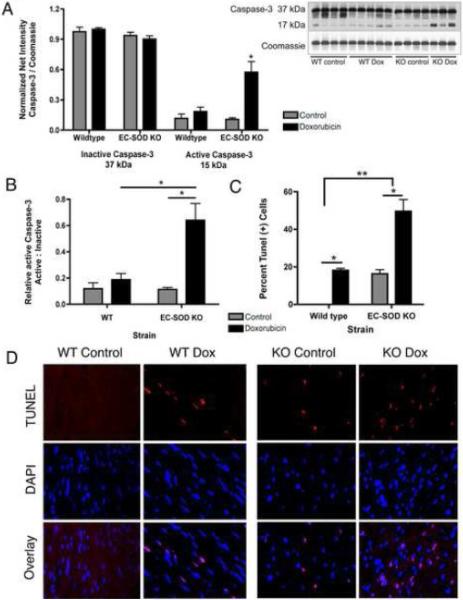
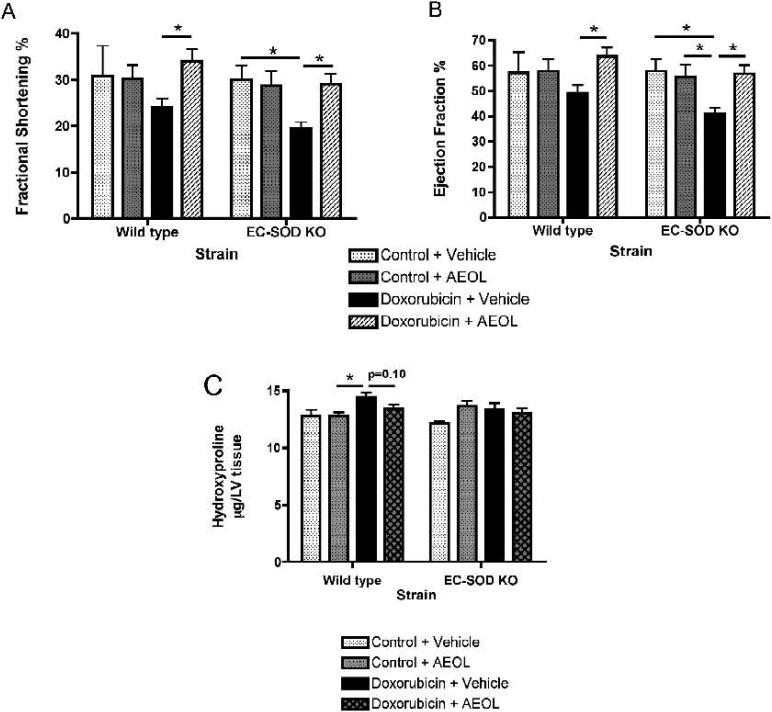
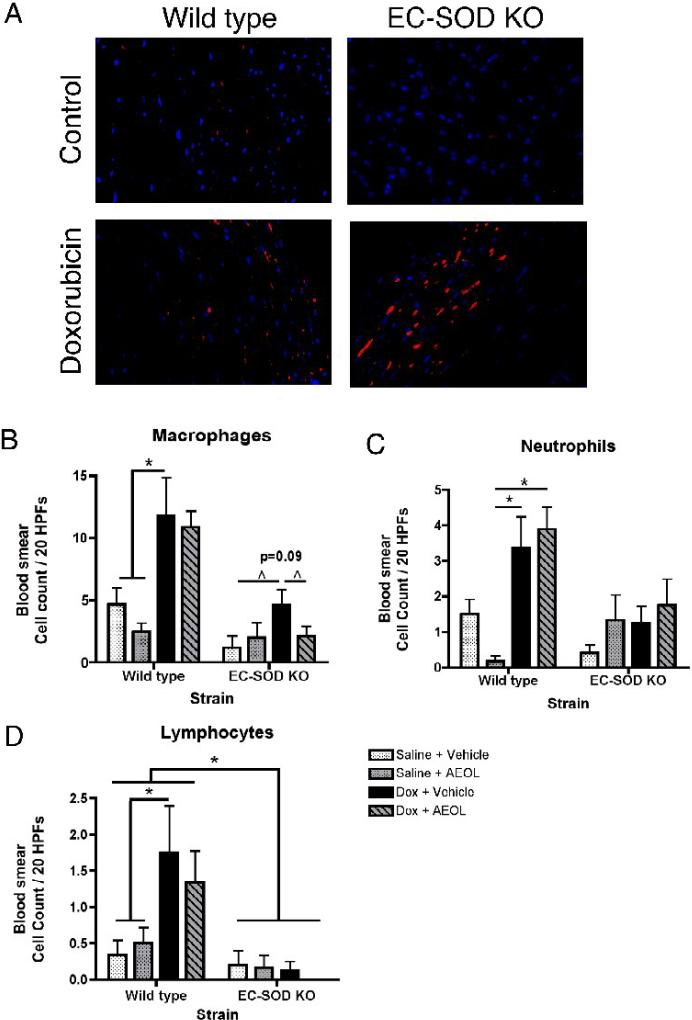
Extracellular superoxide dismutase (EC-SOD) is an antioxidant that protects the heart from ischemia and the lung from inflammation and fibrosis. The role of cardiac EC-SOD under normal conditions and injury remains unclear. Cardiac toxicity, a common side effect of doxorubicin, involves oxidative stress. We hypothesize that EC-SOD is critical for normal cardiac function and protects the heart from oxidant-induced fibrosis and loss of function. C57BL/6 and EC-SOD-null mice were treated with doxorubicin, 15 mg/kg (i.p.). After 15 days, echocardiography was used to assess cardiac function. Left ventricle (LV) tissue was used to assess fibrosis and inflammation by staining, Western blot, and hydroxyproline analysis. At baseline, EC-SOD-null mice have LV wall thinning and increases in LV end diastolic dimensions compared to wild-type mice but have normal cardiac function. After doxorubicin, EC-SOD-null mice have decreases in fractional shortening not apparent in WT mice. Lack of EC-SOD also leads to increases in myocardial apoptosis and significantly more LV fibrosis and inflammatory cell infiltration. Administration of the metalloporphyrin AEOL 10150 abrogates the loss of cardiac function, and potentially fibrosis, associated with doxorubicin treatment in both wild-type and EC-SOD KO mice. EC-SOD is critical for normal cardiac morphology and protects the heart from oxidant-induced fibrosis, apoptosis, and loss of function. The antioxidant metalloporphyrin AEOL 10150 effectively protects cardiac function from doxorubicin-induced oxidative stress in vivo. These findings identify targets for the use of antioxidant agents in oxidant-induced cardiac fibrosis.
Figures
Similar articles
-
Extracellular superoxide dismutase protects the heart against oxidative stress and hypertrophy after myocardial infarction.Free Radic Biol Med. 2008 Apr 1;44(7):1305-13. doi: 10.1016/j.freeradbiomed.2007.12.007. Epub 2007 Dec 15. Free Radic Biol Med. 2008. PMID: 18206658 Free PMC article.
-
Extracellular superoxide dismutase protects cardiovascular syndecan-1 from oxidative shedding.Free Radic Biol Med. 2011 May 1;50(9):1075-80. doi: 10.1016/j.freeradbiomed.2011.02.014. Epub 2011 Feb 18. Free Radic Biol Med. 2011. PMID: 21334435 Free PMC article.
-
Cathepsin A contributes to left ventricular remodeling by degrading extracellular superoxide dismutase in mice.J Biol Chem. 2020 Sep 4;295(36):12605-12617. doi: 10.1074/jbc.RA120.013488. Epub 2020 Jul 9. J Biol Chem. 2020. PMID: 32647007 Free PMC article.
-
Extracellular superoxide dismutase deficiency exacerbates pressure overload-induced left ventricular hypertrophy and dysfunction.Hypertension. 2008 Jan;51(1):19-25. doi: 10.1161/HYPERTENSIONAHA.107.098186. Epub 2007 Nov 12. Hypertension. 2008. PMID: 17998475 Free PMC article.
-
Cardiac inflammation and fibrosis following chemo/radiation therapy: mechanisms and therapeutic agents.Inflammopharmacology. 2022 Feb;30(1):73-89. doi: 10.1007/s10787-021-00894-9. Epub 2021 Nov 23. Inflammopharmacology. 2022. PMID: 34813027 Review.
Cited by
-
The Molecular Mechanisms of Defective Copper Metabolism in Diabetic Cardiomyopathy.Oxid Med Cell Longev. 2022 Oct 4;2022:5418376. doi: 10.1155/2022/5418376. eCollection 2022. Oxid Med Cell Longev. 2022. PMID: 36238639 Free PMC article. Review.
-
Cardiomyocyte-restricted overexpression of extracellular superoxide dismutase increases nitric oxide bioavailability and reduces infarct size after ischemia/reperfusion.Basic Res Cardiol. 2012 Nov;107(6):305. doi: 10.1007/s00395-012-0305-1. Epub 2012 Oct 26. Basic Res Cardiol. 2012. PMID: 23099819 Free PMC article.
-
Redox imbalance in patients with heart failure and ICD/CRT-D intervention. Can it be an underappreciated and overlooked arrhythmogenic factor? A first preliminary clinical study.Front Physiol. 2023 Nov 8;14:1289587. doi: 10.3389/fphys.2023.1289587. eCollection 2023. Front Physiol. 2023. PMID: 38028798 Free PMC article.
-
Role of superoxide-nitric oxide interactions in the accelerated age-related loss of muscle mass in mice lacking Cu,Zn superoxide dismutase.Aging Cell. 2011 Oct;10(5):749-60. doi: 10.1111/j.1474-9726.2011.00709.x. Epub 2011 May 6. Aging Cell. 2011. PMID: 21443684 Free PMC article.
-
Extracellular superoxide dismutase ameliorates skeletal muscle abnormalities, cachexia, and exercise intolerance in mice with congestive heart failure.Circ Heart Fail. 2014 May;7(3):519-30. doi: 10.1161/CIRCHEARTFAILURE.113.000841. Epub 2014 Feb 12. Circ Heart Fail. 2014. PMID: 24523418 Free PMC article.
References
-
- Chen EP, Bittner HB, Davis RD, Folz RJ, Van Trigt P. Extracellular superoxide dismutase transgene overexpression preserves postischemic myocardial function in isolated murine hearts. Circulation. 1996 Nov 1;94(9 Suppl):II412–7. - PubMed
-
- Fattman CL, Schaefer LM, Oury TD. Extracellular superoxide dismutase in biology and medicine. Free radical biology & medicine. 2003 Aug 1;35(3):236–56. - PubMed
-
- Folz RJ, Crapo JD. Extracellular superoxide dismutase (SOD3): tissue-specific expression, genomic characterization, and computer-assisted sequence analysis of the human EC SOD gene. Genomics. 1994 Jul 1;22(1):162–71. - PubMed
-
- Folz RJ, Guan J, Seldin MF, Oury TD, Enghild JJ, Crapo JD. Mouse extracellular superoxide dismutase: primary structure, tissue-specific gene expression, chromosomal localization, and lung in situ hybridization. American journal of respiratory cell and molecular biology. 1997 Oct;17(4):393–403. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
