

Expanding the repertoire of Modified Vaccinia Ankara-based vaccine vectors via genetic complementation strategies
- PMID: 19421328
- PMCID: PMC2674217
- DOI: 10.1371/journal.pone.0005445
Expanding the repertoire of Modified Vaccinia Ankara-based vaccine vectors via genetic complementation strategies
Abstract
Background: Modified Vaccinia virus Ankara (MVA) is a safe, highly attenuated orthopoxvirus that is being developed as a recombinant vaccine vector for immunization against a number of infectious diseases and cancers. However, the expression by MVA vectors of large numbers of poxvirus antigens, which display immunodominance over vectored antigens-of-interest for the priming of T cell responses, and the induction of vector-neutralizing antibodies, which curtail the efficacy of subsequent booster immunizations, remain as significant impediments to the overall utility of such vaccines. Thus, genetic approaches that enable the derivation of MVA vectors that are antigenically less complex may allow for rational improvement of MVA-based vaccines.
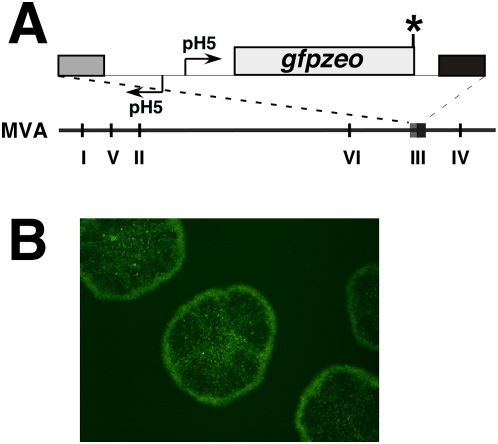
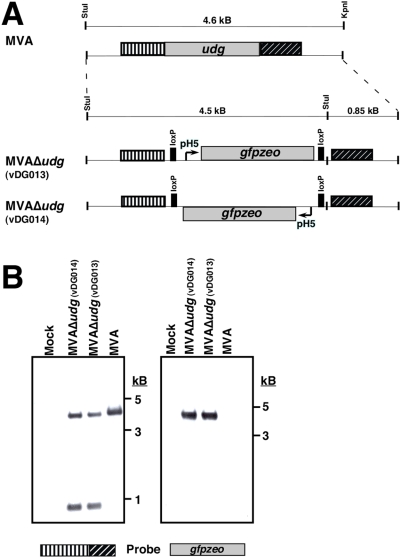
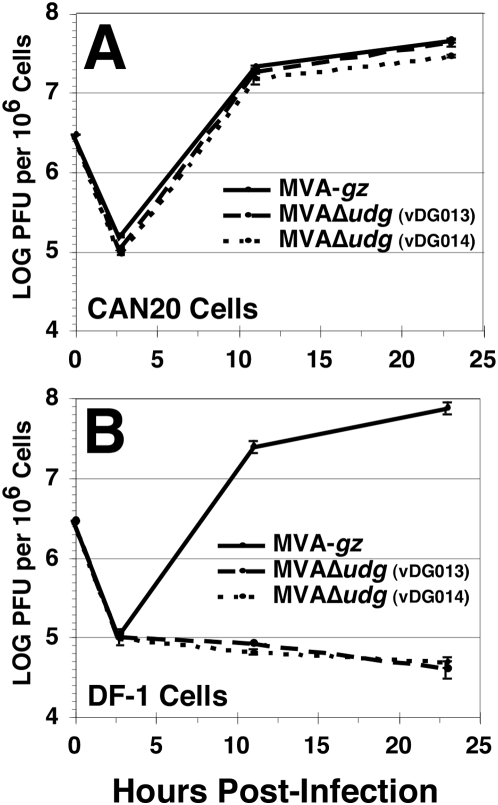
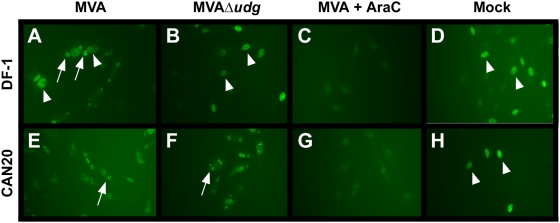
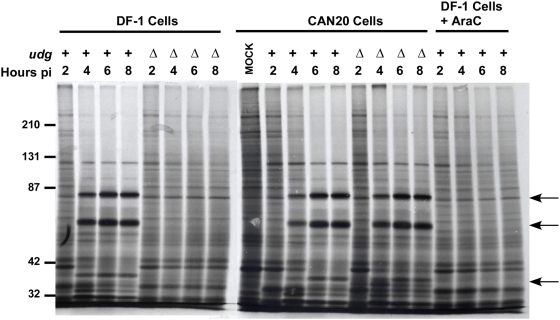
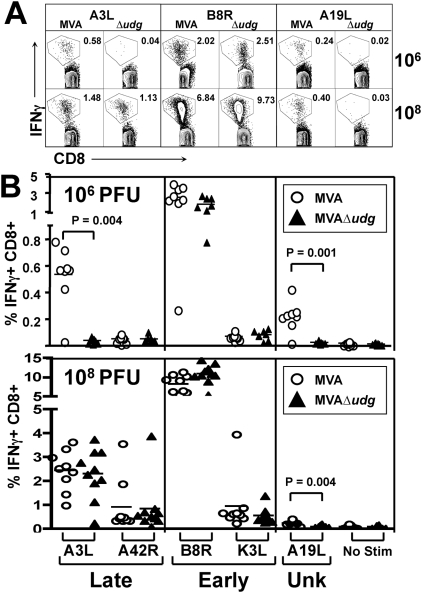
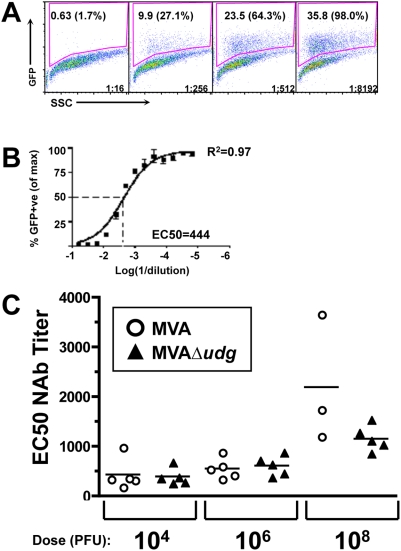
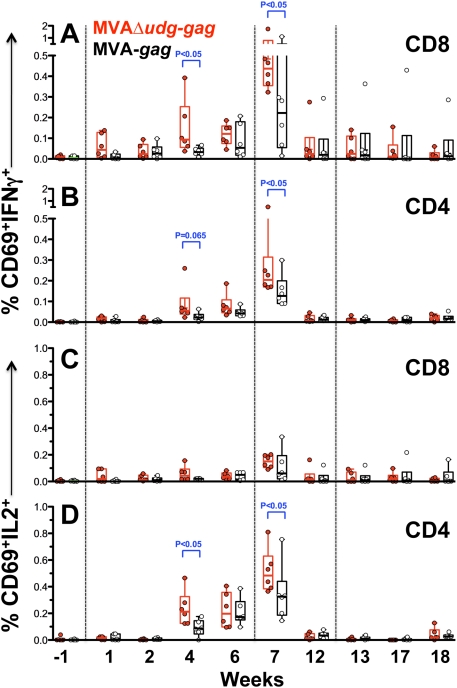
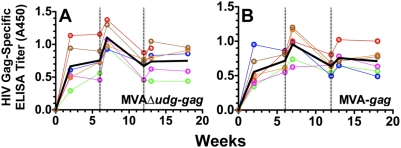
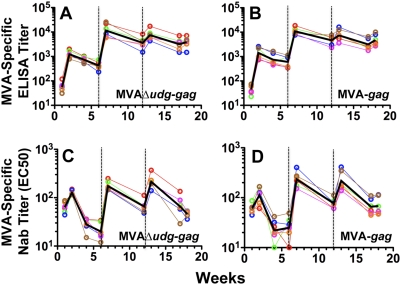
Principal findings: We have developed a genetic complementation system that enables the deletion of essential viral genes from the MVA genome, thereby allowing us to generate MVA vaccine vectors that are antigenically less complex. Using this system, we deleted the essential uracil-DNA-glycosylase (udg) gene from MVA and propagated this otherwise replication-defective variant on a complementing cell line that constitutively expresses the poxvirus udg gene and that was derived from a newly identified continuous cell line that is permissive for growth of wild type MVA. The resulting virus, MVADeltaudg, does not replicate its DNA genome or express late viral gene products during infection of non-complementing cells in culture. As proof-of-concept for immunological 'focusing', we demonstrate that immunization of mice with MVADeltaudg elicits CD8+ T cell responses that are directed against a restricted repertoire of vector antigens, as compared to immunization with parental MVA. Immunization of rhesus macaques with MVADeltaudg-gag, a udg(-) recombinant virus that expresses an HIV subtype-B consensus gag transgene, elicited significantly higher frequencies of Gag-specific CD8 and CD4 T cells following both primary (2-4-fold) and booster (2-fold) immunizations as compared to the udg(+) control virus MVA-gag, as determined by intracellular cytokine assay. In contrast, levels of HIV Gag-specific antibodies were elicited similarly in macaques following immunization with MVADeltaudg-gag and MVA-gag. Furthermore, both udg(-) and udg(+) MVA vectors induced comparatively similar titers of MVA-specific neutralizing antibody responses following immunization of mice (over a 4-log range: 10(4)-10(8) PFU) and rhesus macaques. These results suggest that the generation of MVA-specific neutralizing antibody responses are largely driven by input MVA antigens, rather than those that are synthesized de novo during infection, and that the processes governing the generation of antiviral antibody responses are more readily saturated by viral antigen than are those that elicit CD8+ T cell responses.
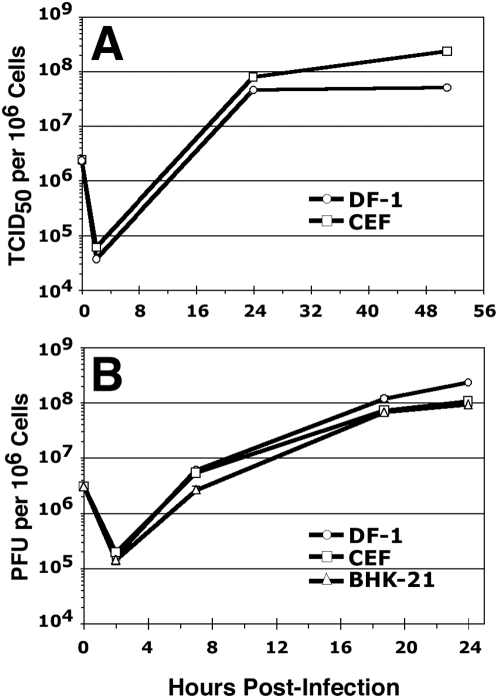
Significance: Our identification of a spontaneously-immortalized (but not transformed) chicken embryo fibroblast cell line (DF-1) that is fully permissive for MVA growth and that can be engineered to stably express MVA genes provides the basis for a genetic system for MVA. DF-1 cells (and derivatives thereof) constitute viable alternatives, for the manufacture of MVA-based vaccines, to primary CEFs -- the conventional cell substrate for MVA vaccines that is not amenable to genetic complementation strategies due to these cells' finite lifespan in culture. The establishment of a genetic system for MVA, as illustrated here to allow udg deletion, enables the generation of novel replication-defective MVA mutants and expands the repertoire of genetic viral variants that can now be explored as improved vaccine vectors.
Conflict of interest statement
Figures
Similar articles
-
Deletion of specific immune-modulatory genes from modified vaccinia virus Ankara-based HIV vaccines engenders improved immunogenicity in rhesus macaques.J Virol. 2012 Dec;86(23):12605-15. doi: 10.1128/JVI.00246-12. Epub 2012 Sep 12. J Virol. 2012. PMID: 22973033 Free PMC article.
-
Deletion of the vaccinia virus N2L gene encoding an inhibitor of IRF3 improves the immunogenicity of modified vaccinia virus Ankara expressing HIV-1 antigens.J Virol. 2014 Mar;88(6):3392-410. doi: 10.1128/JVI.02723-13. Epub 2014 Jan 3. J Virol. 2014. PMID: 24390336 Free PMC article.
-
Potent Anti-hepatitis C Virus (HCV) T Cell Immune Responses Induced in Mice Vaccinated with DNA-Launched RNA Replicons and Modified Vaccinia Virus Ankara-HCV.J Virol. 2019 Mar 21;93(7):e00055-19. doi: 10.1128/JVI.00055-19. Print 2019 Apr 1. J Virol. 2019. PMID: 30674625 Free PMC article.
-
Development of a DNA-MVA/HIVA vaccine for Kenya.Vaccine. 2002 May 6;20(15):1995-8. doi: 10.1016/s0264-410x(02)00085-3. Vaccine. 2002. PMID: 11983261 Review.
-
Modified Vaccinia Virus Ankara: History, Value in Basic Research, and Current Perspectives for Vaccine Development.Adv Virus Res. 2017;97:187-243. doi: 10.1016/bs.aivir.2016.07.001. Epub 2016 Aug 1. Adv Virus Res. 2017. PMID: 28057259 Free PMC article. Review.
Cited by
-
Deletion of the vaccinia virus gene A46R, encoding for an inhibitor of TLR signalling, is an effective approach to enhance the immunogenicity in mice of the HIV/AIDS vaccine candidate NYVAC-C.PLoS One. 2013 Sep 17;8(9):e74831. doi: 10.1371/journal.pone.0074831. eCollection 2013. PLoS One. 2013. PMID: 24069354 Free PMC article.
-
Gene therapy for lung neoplasms.Clin Chest Med. 2011 Dec;32(4):865-85. doi: 10.1016/j.ccm.2011.08.006. Epub 2011 Oct 7. Clin Chest Med. 2011. PMID: 22054892 Free PMC article. Review.
-
Improving Adaptive and Memory Immune Responses of an HIV/AIDS Vaccine Candidate MVA-B by Deletion of Vaccinia Virus Genes (C6L and K7R) Blocking Interferon Signaling Pathways.PLoS One. 2013 Jun 27;8(6):e66894. doi: 10.1371/journal.pone.0066894. Print 2013. PLoS One. 2013. PMID: 23826170 Free PMC article.
-
The evolution of poxvirus vaccines.Viruses. 2015 Apr 7;7(4):1726-803. doi: 10.3390/v7041726. Viruses. 2015. PMID: 25853483 Free PMC article. Review.
-
New approaches to design HIV-1 T-cell vaccines.Curr Opin HIV AIDS. 2010 Sep;5(5):368-76. doi: 10.1097/COH.0b013e32833d2cc0. Curr Opin HIV AIDS. 2010. PMID: 20978376 Free PMC article. Review.
References
-
- Mayr A, Hochstein-Mintzel V, Stickl H. Abstammung, eigenschaften und verwendung des attenuierten vaccinia-stammes MVA [Passage history, properties, and applicability of the attenuated vaccinia virus strain MVA]. Infection. 1975;3:6–14.
-
- Meyer H, Sutter G, Mayr A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. Journal of General Virology. 1991;72:1031–1038. - PubMed
-
- Antoine G, Scheiflinger F, Dorner F, Falkner FG. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology. 1998;244:365–396. - PubMed
-
- Blanchard TJ, Alcami A, Andrea P, Smith GL. Modified vaccinia virus Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: implications for use as a human vaccine. Journal of General Virology. 1998;79:1159–1167. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
