

Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells
- PMID: 19329998
- PMCID: PMC3566772
- DOI: 10.1038/nbt.1533
Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells
Erratum in
- Nat Biotechnol. 2009 May;27(5):485
Abstract
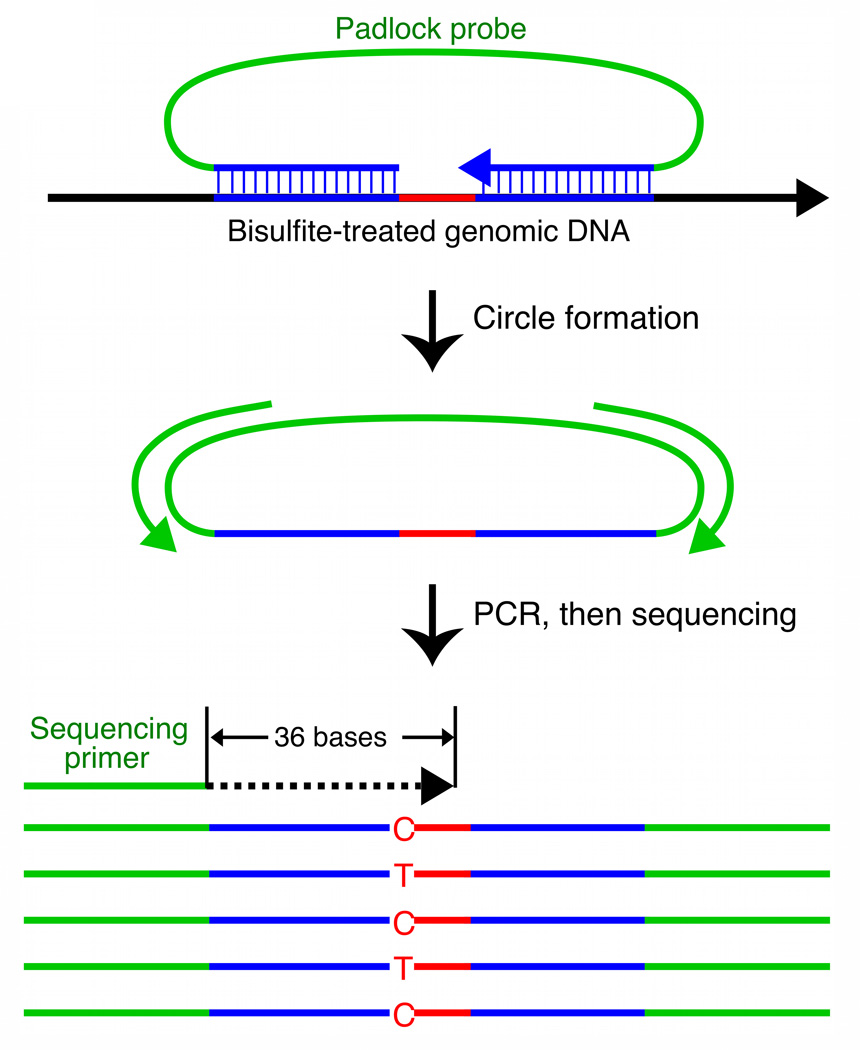
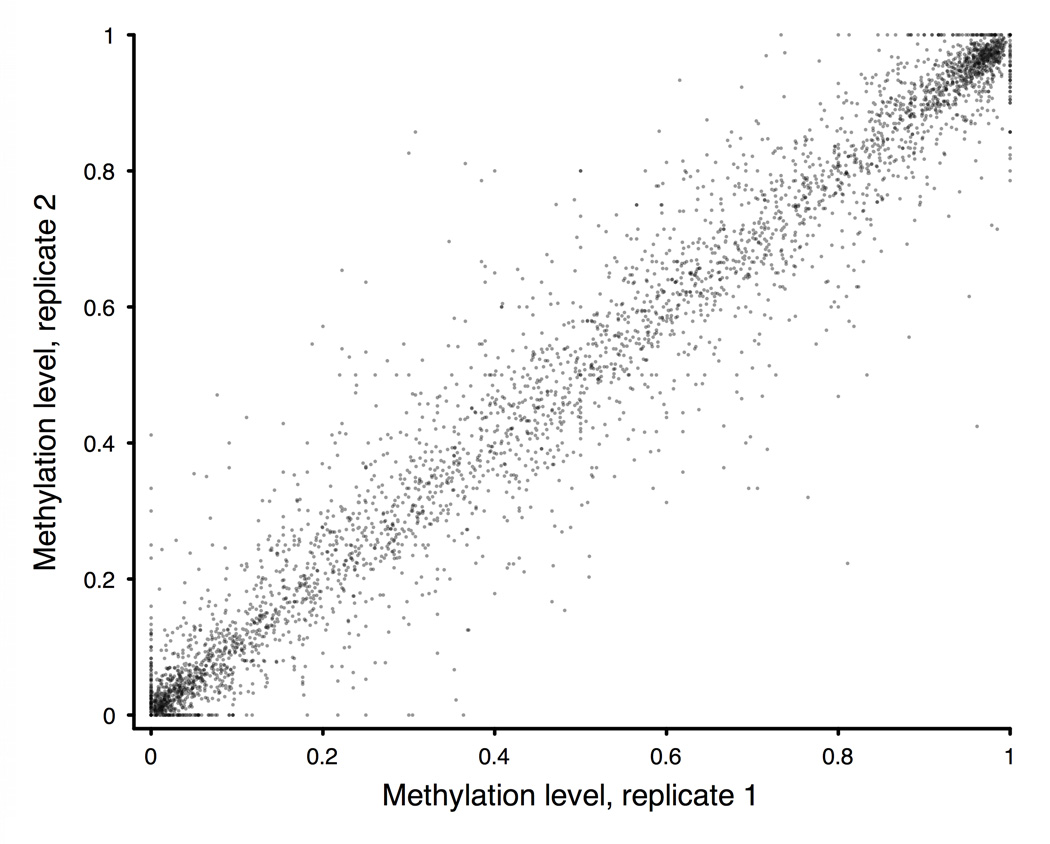
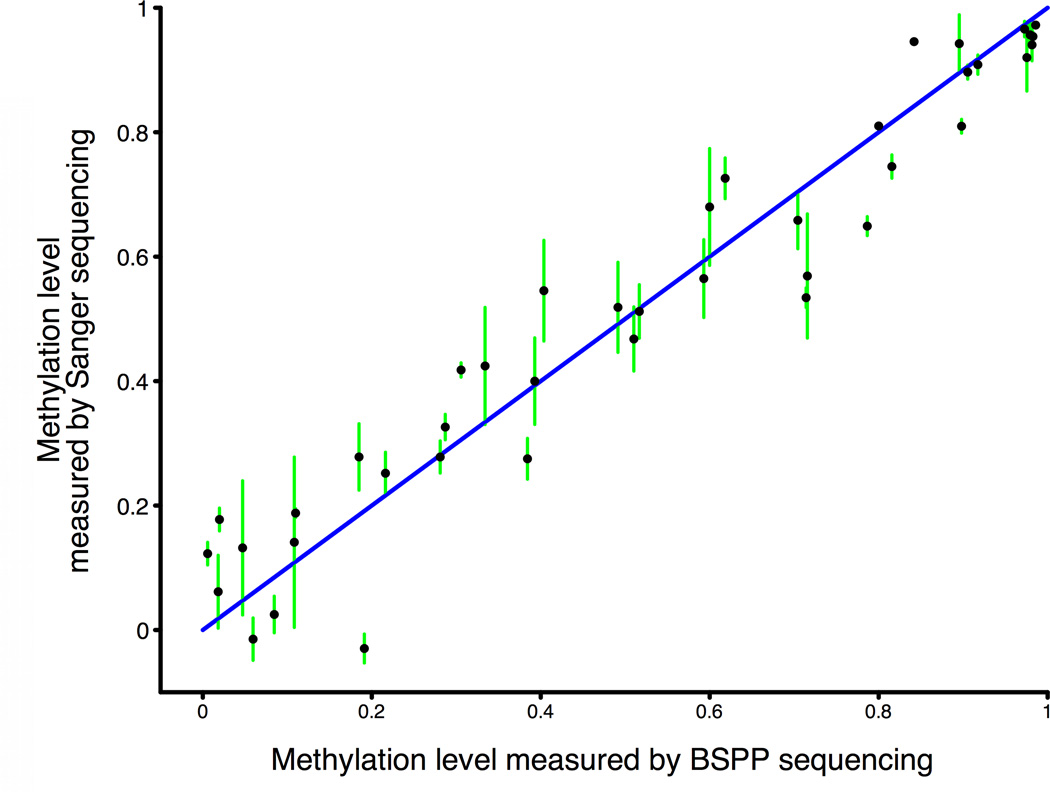
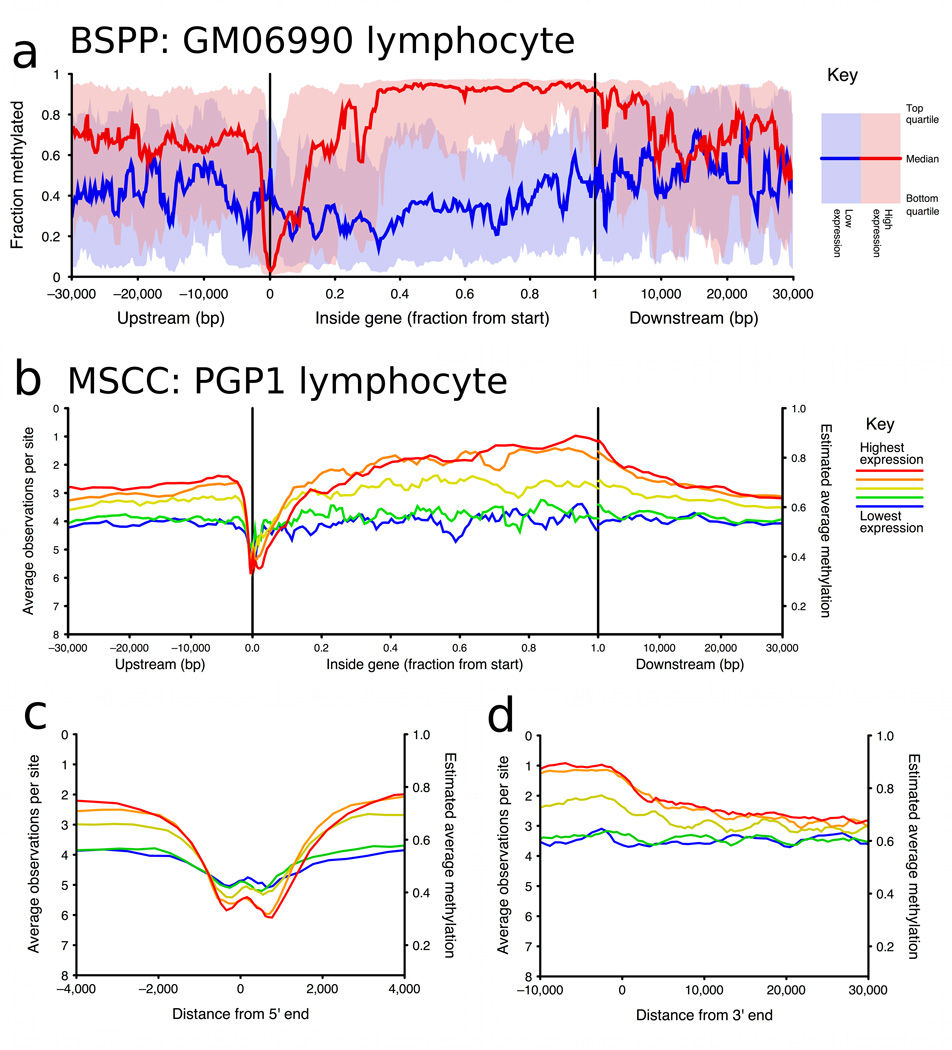
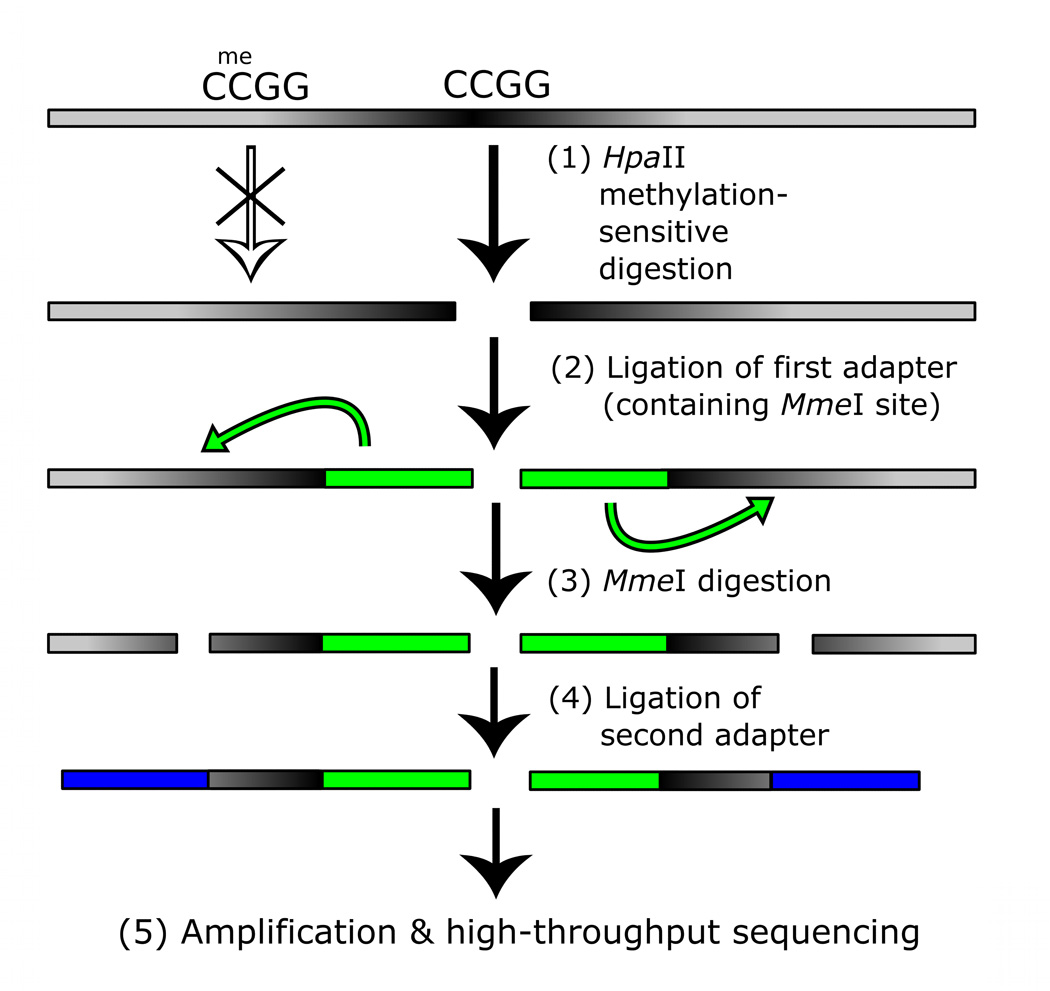
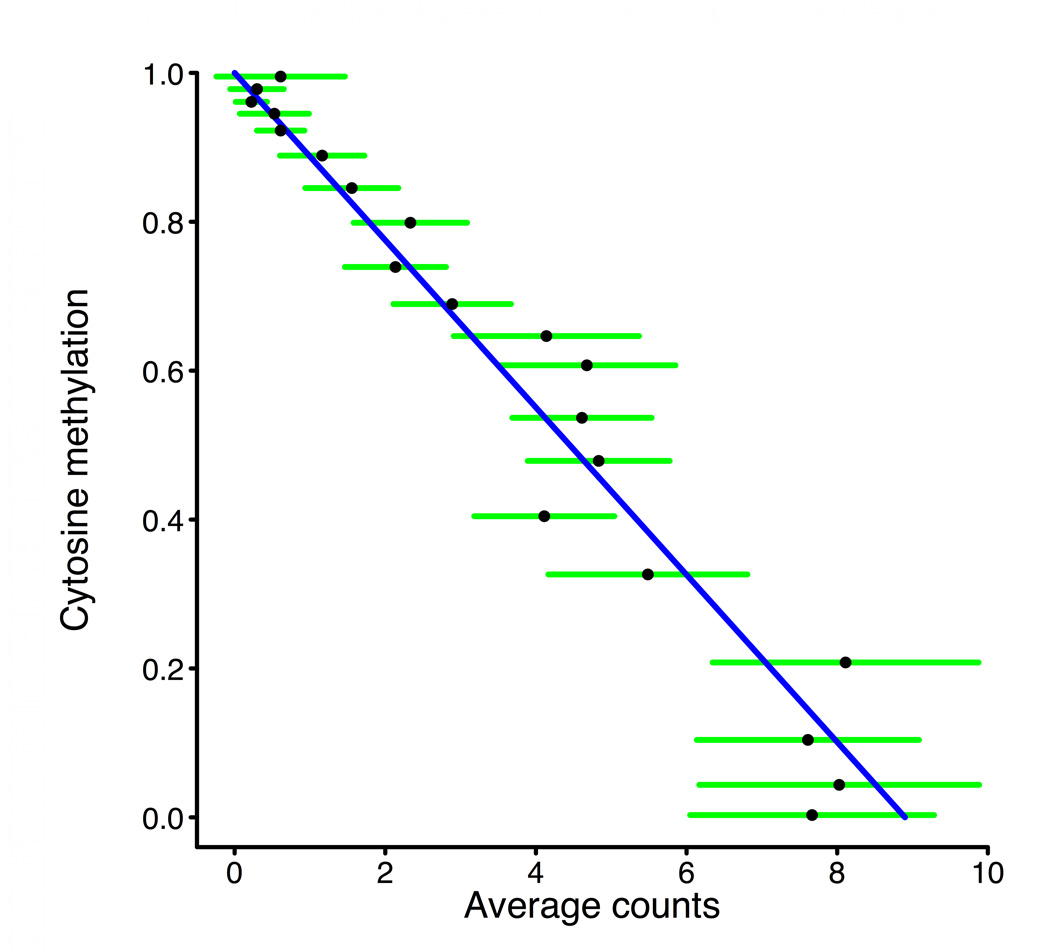
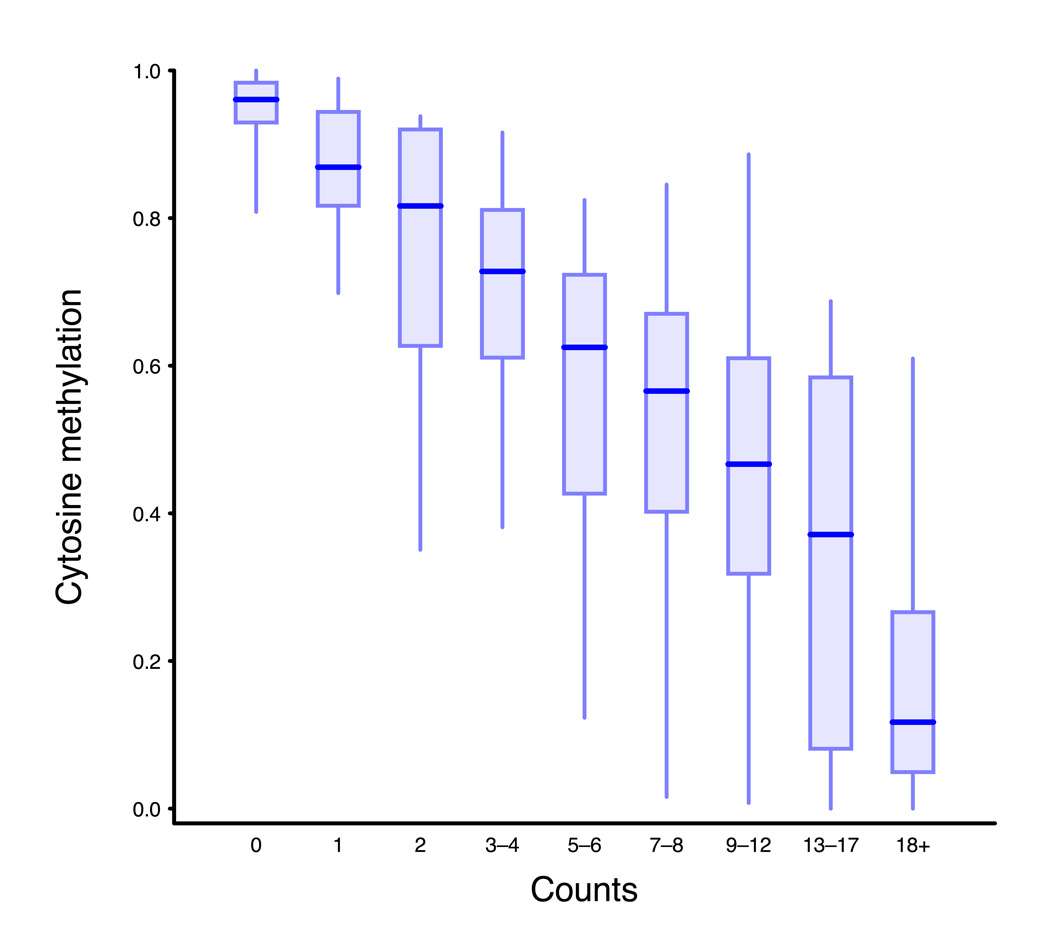
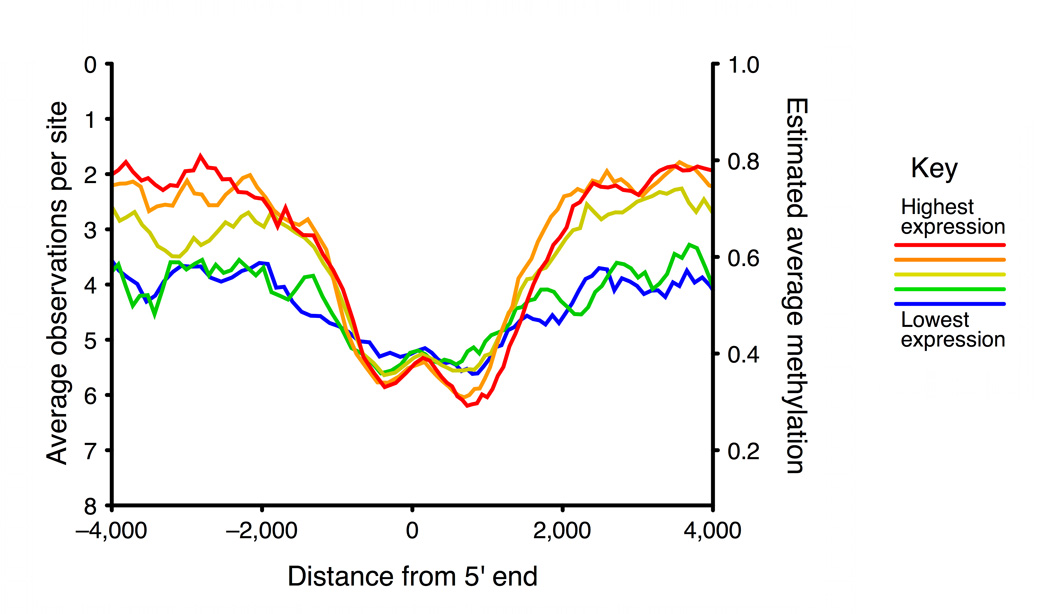
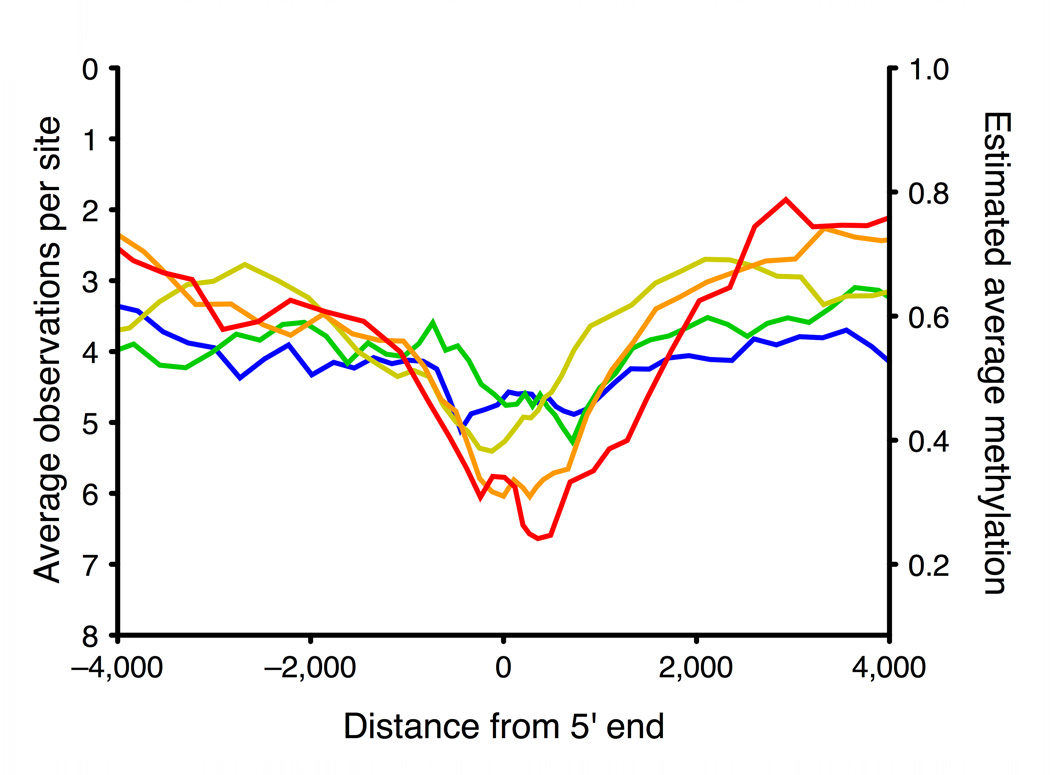
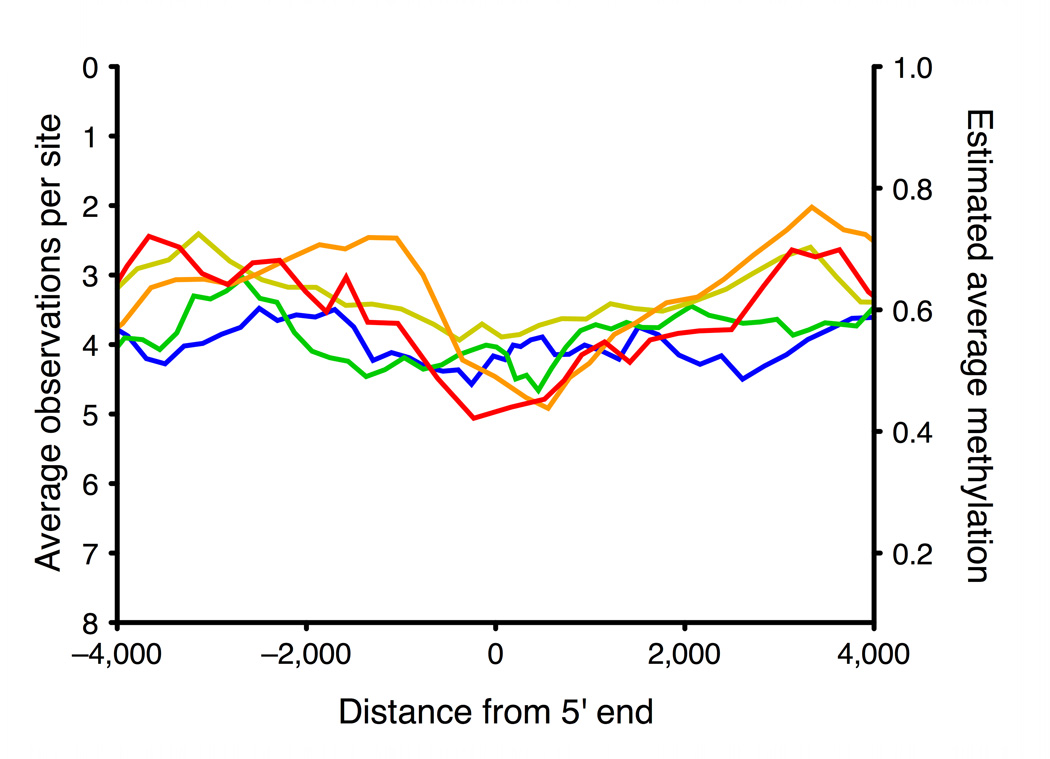
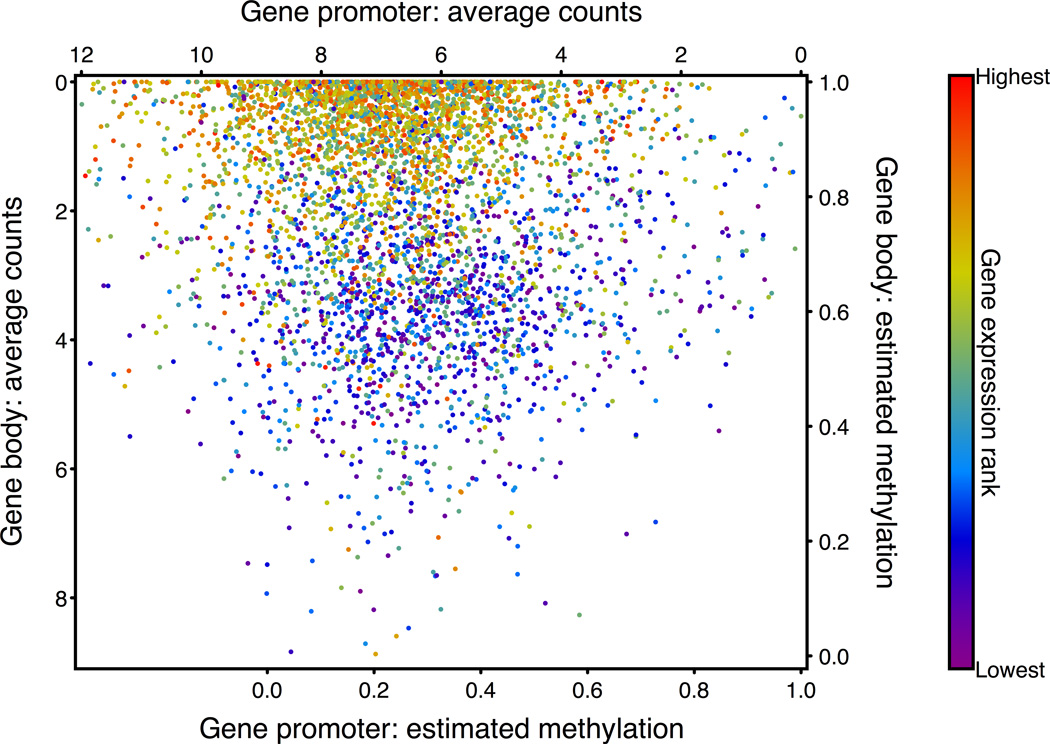
Studies of epigenetic modifications would benefit from improved methods for high-throughput methylation profiling. We introduce two complementary approaches that use next-generation sequencing technology to detect cytosine methylation. In the first method, we designed approximately 10,000 bisulfite padlock probes to profile approximately 7,000 CpG locations distributed over the ENCODE pilot project regions and applied them to human B-lymphocytes, fibroblasts and induced pluripotent stem cells. This unbiased choice of targets takes advantage of existing expression and chromatin immunoprecipitation data and enabled us to observe a pattern of low promoter methylation and high gene-body methylation in highly expressed genes. The second method, methyl-sensitive cut counting, generated nontargeted genome-scale data for approximately 1.4 million HpaII sites in the DNA of B-lymphocytes and confirmed that gene-body methylation in highly expressed genes is a consistent phenomenon throughout the human genome. Our observations highlight the usefulness of techniques that are not inherently or intentionally biased towards particular subsets like CpG islands or promoter regions.
Figures
Comment in
-
Locking in on the human methylome.Nat Biotechnol. 2009 Apr;27(4):341-2. doi: 10.1038/nbt0409-341. Nat Biotechnol. 2009. PMID: 19352369 No abstract available.
Similar articles
-
Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming.Nat Biotechnol. 2009 Apr;27(4):353-60. doi: 10.1038/nbt.1530. Epub 2009 Mar 29. Nat Biotechnol. 2009. PMID: 19330000 Free PMC article.
-
Methodological aspects of whole-genome bisulfite sequencing analysis.Brief Bioinform. 2015 May;16(3):369-79. doi: 10.1093/bib/bbu016. Epub 2014 May 27. Brief Bioinform. 2015. PMID: 24867940 Review.
-
Targeted DNA methylation analysis by next-generation sequencing.J Vis Exp. 2015 Feb 24;(96):52488. doi: 10.3791/52488. J Vis Exp. 2015. PMID: 25741966 Free PMC article.
-
Application of microdroplet PCR for large-scale targeted bisulfite sequencing.Genome Res. 2011 Oct;21(10):1738-45. doi: 10.1101/gr.116863.110. Epub 2011 Jul 14. Genome Res. 2011. PMID: 21757609 Free PMC article.
-
Genome-wide analysis of DNA methylation patterns.Development. 2007 Nov;134(22):3959-65. doi: 10.1242/dev.001131. Epub 2007 Oct 10. Development. 2007. PMID: 17928417 Review.
Cited by
-
Environmental exposures influence multigenerational epigenetic transmission.Clin Epigenetics. 2024 Oct 17;16(1):145. doi: 10.1186/s13148-024-01762-3. Clin Epigenetics. 2024. PMID: 39420431 Free PMC article. Review.
-
DNA methylation profiling identifies TBKBP1 as potent amplifier of cytotoxic activity in CMV-specific human CD8+ T cells.PLoS Pathog. 2024 Sep 26;20(9):e1012581. doi: 10.1371/journal.ppat.1012581. eCollection 2024 Sep. PLoS Pathog. 2024. PMID: 39325839 Free PMC article.
-
Genetic variation drives cancer cell adaptation to ECM stiffness.Proc Natl Acad Sci U S A. 2024 Sep 24;121(39):e2403062121. doi: 10.1073/pnas.2403062121. Epub 2024 Sep 20. Proc Natl Acad Sci U S A. 2024. PMID: 39302966 Free PMC article.
-
Triphenyl Phosphate Alters Methyltransferase Expression and Induces Genome-Wide Aberrant DNA Methylation in Zebrafish Larvae.Chem Res Toxicol. 2024 Sep 16;37(9):1549-1561. doi: 10.1021/acs.chemrestox.4c00223. Epub 2024 Aug 29. Chem Res Toxicol. 2024. PMID: 39205618 Free PMC article.
-
Dual Regulation Mechanism of Obesity: DNA Methylation and Intestinal Flora.Biomedicines. 2024 Jul 23;12(8):1633. doi: 10.3390/biomedicines12081633. Biomedicines. 2024. PMID: 39200098 Free PMC article. Review.
References
-
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. - PubMed
-
- Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. - PubMed
-
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. - PubMed
-
- Jiang YH, Bressler J, Beaudet AL. Epigenetics and human disease. Annu Rev Genomics Hum Genet. 2004;5:479–510. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
