

Protein replacement therapy and gene transfer in canine models of hemophilia A, hemophilia B, von willebrand disease, and factor VII deficiency
- PMID: 19293459
- PMCID: PMC3101868
- DOI: 10.1093/ilar.50.2.144
Protein replacement therapy and gene transfer in canine models of hemophilia A, hemophilia B, von willebrand disease, and factor VII deficiency
Abstract
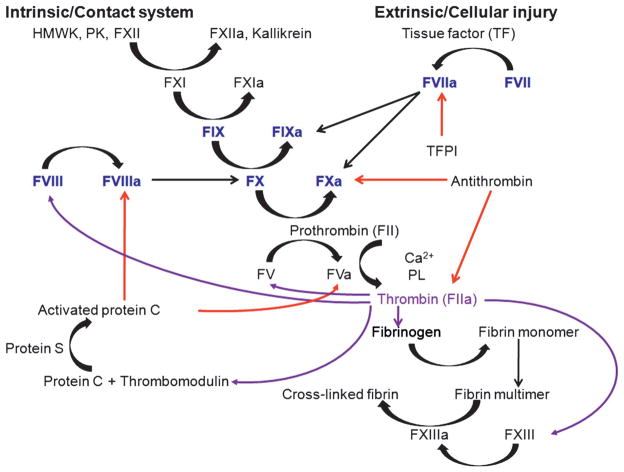
Dogs with hemophilia A, hemophilia B, von Willebrand disease (VWD), and factor VII deficiency faithfully recapitulate the severe bleeding phenotype that occurs in humans with these disorders. The first rational approach to diagnosing these bleeding disorders became possible with the development of reliable assays in the 1940s through research that used these dogs. For the next 60 years, treatment consisted of replacement of the associated missing or dysfunctional protein, first with plasma-derived products and subsequently with recombinant products. Research has consistently shown that replacement products that are safe and efficacious in these dogs prove to be safe and efficacious in humans. But these highly effective products require repeated administration and are limited in supply and expensive; in addition, plasma-derived products have transmitted bloodborne pathogens. Recombinant proteins have all but eliminated inadvertent transmission of bloodborne pathogens, but the other limitations persist. Thus, gene therapy is an attractive alternative strategy in these monogenic disorders and has been actively pursued since the early 1990s. To date, several modalities of gene transfer in canine hemophilia have proven to be safe, produced easily detectable levels of transgene products in plasma that have persisted for years in association with reduced bleeding, and correctly predicted the vector dose required in a human hemophilia B liver-based trial. Very recently, however, researchers have identified an immune response to adeno-associated viral gene transfer vector capsid proteins in a human liver-based trial that was not present in preclinical testing in rodents, dogs, or nonhuman primates. This article provides a review of the strengths and limitations of canine hemophilia, VWD, and factor VII deficiency models and of their historical and current role in the development of improved therapy for humans with these inherited bleeding disorders.
Figures
Similar articles
-
Hereditary hemorrhagic disorders.Rev Invest Clin. 1994 Apr;Suppl:134-43. Rev Invest Clin. 1994. PMID: 7886297 Review. No abstract available.
-
Animal models of hemophilia and related bleeding disorders.Semin Hematol. 2013 Apr;50(2):175-84. doi: 10.1053/j.seminhematol.2013.03.023. Semin Hematol. 2013. PMID: 23956467 Free PMC article. Review.
-
Canine models of inherited bleeding disorders in the development of coagulation assays, novel protein replacement and gene therapies.J Thromb Haemost. 2016 May;14(5):894-905. doi: 10.1111/jth.13301. Epub 2016 Apr 5. J Thromb Haemost. 2016. PMID: 26924758 Review.
-
Advances toward gene therapy for hemophilia at the millennium.Hum Gene Ther. 1999 Sep 1;10(13):2091-107. doi: 10.1089/10430349950017095. Hum Gene Ther. 1999. PMID: 10498242 Review.
-
Effect of recombinant factor VIIa on the hemostatic defect in dogs with hemophilia A, hemophilia B, and von Willebrand disease.Proc Natl Acad Sci U S A. 1989 Feb;86(4):1382-6. doi: 10.1073/pnas.86.4.1382. Proc Natl Acad Sci U S A. 1989. PMID: 2784006 Free PMC article.
Cited by
-
ARFI ultrasound monitoring of hemorrhage and hemostasis in vivo in canine von Willebrand disease and hemophilia.Ultrasound Med Biol. 2011 Dec;37(12):2126-32. doi: 10.1016/j.ultrasmedbio.2011.09.006. Epub 2011 Oct 26. Ultrasound Med Biol. 2011. PMID: 22033127 Free PMC article.
-
Sensitivity of whole blood clotting time and activated partial thromboplastin time for factor IX: relevance to gene therapy and determination of post-transfusion elimination time of canine factor IX in hemophilia B dogs.J Thromb Haemost. 2012 Mar;10(3):474-6. doi: 10.1111/j.1538-7836.2011.04613.x. J Thromb Haemost. 2012. PMID: 22482117 Free PMC article. No abstract available.
-
Current animal models of hemophilia: the state of the art.Thromb J. 2016 Oct 4;14(Suppl 1):22. doi: 10.1186/s12959-016-0106-0. eCollection 2016. Thromb J. 2016. PMID: 27766048 Free PMC article. Review.
-
Phenotypic correction of hemophilia A in sheep by postnatal intraperitoneal transplantation of FVIII-expressing MSC.Exp Hematol. 2011 Dec;39(12):1124-1135.e4. doi: 10.1016/j.exphem.2011.09.001. Epub 2011 Sep 8. Exp Hematol. 2011. PMID: 21906573 Free PMC article.
-
Porcine and canine von Willebrand factor and von Willebrand disease: hemostasis, thrombosis, and atherosclerosis studies.Thrombosis. 2010;2010:461238. doi: 10.1155/2010/461238. Epub 2011 Feb 7. Thrombosis. 2010. PMID: 22091368 Free PMC article.
References
-
- Aledort LM, DiMichele DM. Inhibitors occur more frequently in Africo-Americans and Latino hæmophiliacs. Hæmophilia. 1998;80:779–783. - PubMed
-
- Antonarakis SE, Rossiter JP, Young M, Horst J, de Moerloose P, Sommer SS, Ketterling RP, Kazazian HH, Jr, Negrier C, Vinciguerra C, Gitschier J, Goossens M, Girodon E, Ghanem N, Plassa F, Lavergne JM, Vidaud M, Costa JM, Laurian Y, Lin S-W, Lin S-R, Shen M-C, Lillicrap D, Taylor SAM, Windsor S, Valleix SV, Nafa K, Sultan Y, Delpech M, Vnencak-Jones C, Phillips JA, III, Ljung RCR, Koumbarelis E, Gialeraki A, Mandalaki T, Jenkins PV, Collins PW, Pasi KJ, Goodeve A, Peake I, Preston FE, Schwartz M, Scheibel E, Ingerslev J, Cooper DN, Millar DS, Kakkar VV, Giannelli F, Naylor JA, Tizzano EF, Baiget M, Domenech M, Altisent C, Tusell J, Beneyto M, Lorenzo JI, Gaucher C, Mazurier C, Peerlinck GMK, Matthijs K, Cassiman JJ, Vermylen J, Mori PG, Acquila M, Caprino D, Inaba H. Factor VIII gene inversions in severe hemophilia A: Results of an international consortium study. Blood. 1995;86:2206–2212. - PubMed
-
- Arruda VR. Toward gene therapy for hemophilia A with novel adenoviral vectors: Successes and limitations in canine models. J Thromb Hæmost. 2006;4:1215–1217. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
