

Solvent accessible surface area approximations for rapid and accurate protein structure prediction
- PMID: 19234730
- PMCID: PMC2712621
- DOI: 10.1007/s00894-009-0454-9
Solvent accessible surface area approximations for rapid and accurate protein structure prediction
Abstract
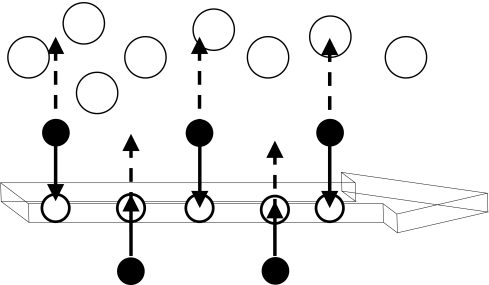
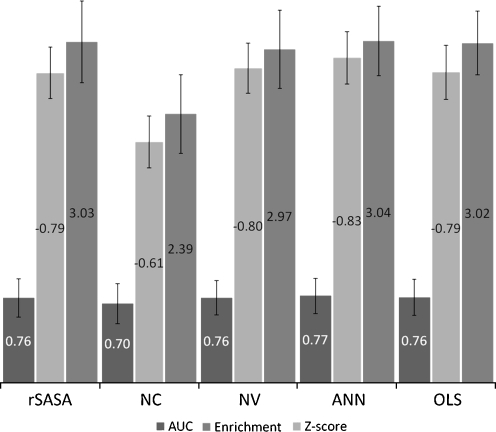
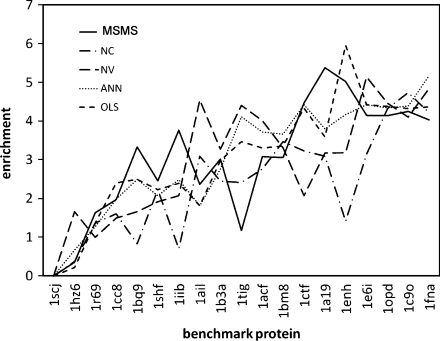
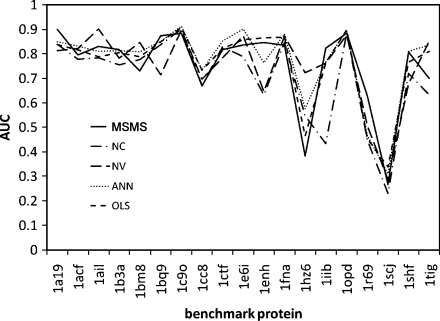
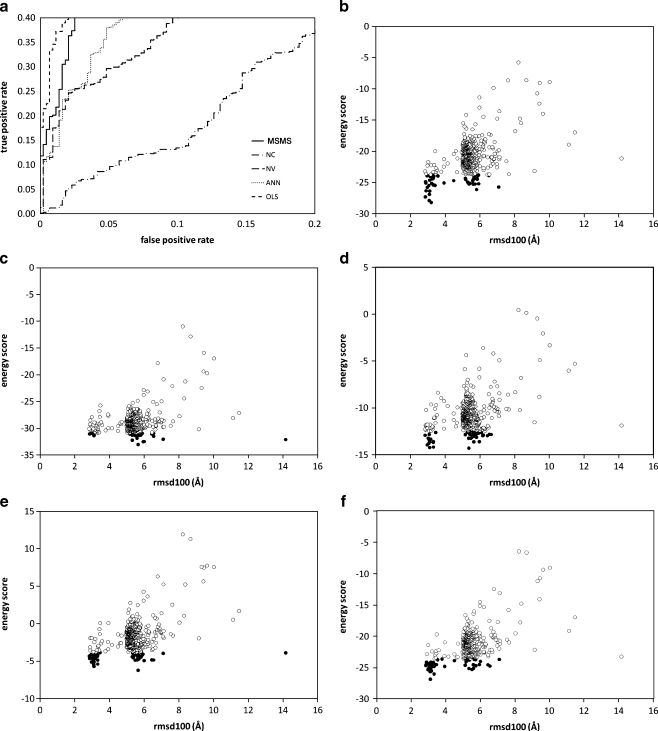
The burial of hydrophobic amino acids in the protein core is a driving force in protein folding. The extent to which an amino acid interacts with the solvent and the protein core is naturally proportional to the surface area exposed to these environments. However, an accurate calculation of the solvent-accessible surface area (SASA), a geometric measure of this exposure, is numerically demanding as it is not pair-wise decomposable. Furthermore, it depends on a full-atom representation of the molecule. This manuscript introduces a series of four SASA approximations of increasing computational complexity and accuracy as well as knowledge-based environment free energy potentials based on these SASA approximations. Their ability to distinguish correctly from incorrectly folded protein models is assessed to balance speed and accuracy for protein structure prediction. We find the newly developed "Neighbor Vector" algorithm provides the most optimal balance of accurate yet rapid exposure measures.
Figures
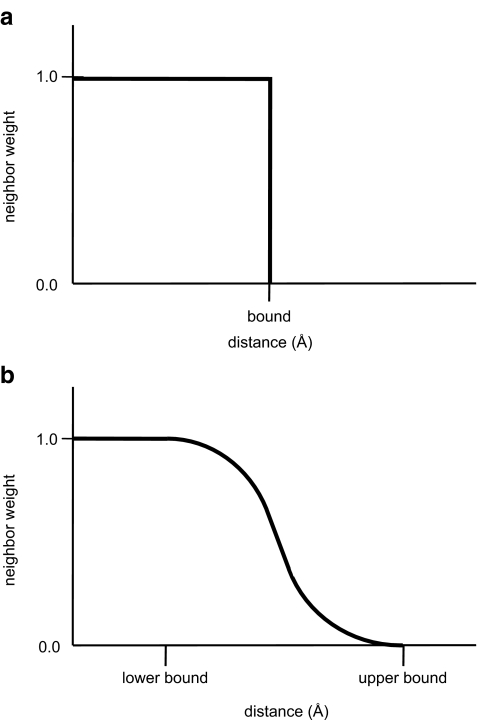
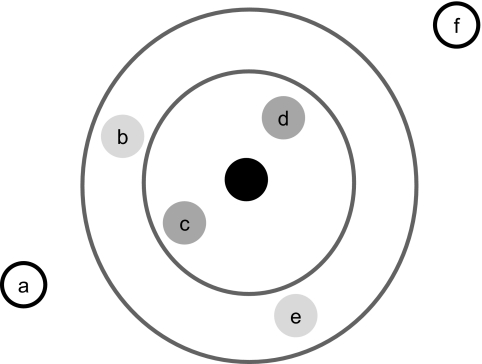
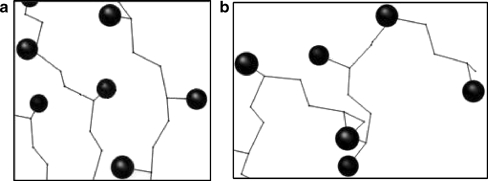
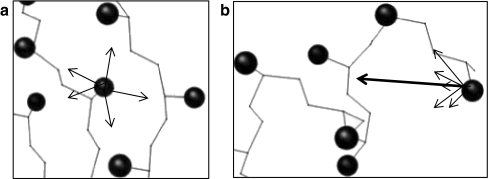


Similar articles
-
A hydrophobic spine stabilizes a surface-exposed α-helix according to analysis of the solvent-accessible surface area.BMC Bioinformatics. 2016 Dec 22;17(Suppl 19):503. doi: 10.1186/s12859-016-1368-z. BMC Bioinformatics. 2016. PMID: 28155647 Free PMC article.
-
Efficient approximate all-atom solvent accessible surface area method parameterized for folded and denatured protein conformations.J Comput Chem. 2004 Jun;25(8):1005-14. doi: 10.1002/jcc.20026. J Comput Chem. 2004. PMID: 15067676
-
A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states.Curr Protein Pept Sci. 2014;15(5):456-76. doi: 10.2174/1389203715666140327114232. Curr Protein Pept Sci. 2014. PMID: 24678666 Review.
-
Quantification of protein surfaces, volumes and atom-atom contacts using a constrained Voronoi procedure.Bioinformatics. 2002 Oct;18(10):1365-73. doi: 10.1093/bioinformatics/18.10.1365. Bioinformatics. 2002. PMID: 12376381
-
Atom depth in protein structure and function.Trends Biochem Sci. 2003 Nov;28(11):593-7. doi: 10.1016/j.tibs.2003.09.004. Trends Biochem Sci. 2003. PMID: 14607089 Review.
Cited by
-
Screening of phytoconstituents from Bacopa monnieri (L.) Pennell and Mucuna pruriens (L.) DC. to identify potential inhibitors against Cerebroside sulfotransferase.PLoS One. 2024 Oct 24;19(10):e0307374. doi: 10.1371/journal.pone.0307374. eCollection 2024. PLoS One. 2024. PMID: 39446901 Free PMC article.
-
Piperine's potential in treating polycystic ovarian syndrome explored through in-silico docking.Sci Rep. 2024 Sep 18;14(1):21834. doi: 10.1038/s41598-024-72800-6. Sci Rep. 2024. PMID: 39294254 Free PMC article.
-
Biophysical and structural considerations for protein sequence evolution.BMC Evol Biol. 2011 Dec 16;11:361. doi: 10.1186/1471-2148-11-361. BMC Evol Biol. 2011. PMID: 22171550 Free PMC article.
-
Inhibition of Monkeypox Virus DNA Polymerase Using Moringa oleifera Phytochemicals: Computational Studies of Drug-Likeness, Molecular Docking, Molecular Dynamics Simulation and Density Functional Theory.Indian J Microbiol. 2024 Sep;64(3):1057-1074. doi: 10.1007/s12088-024-01244-3. Epub 2024 Mar 28. Indian J Microbiol. 2024. PMID: 39282169
-
Mass spectrometry coupled experiments and protein structure modeling methods.Int J Mol Sci. 2013 Oct 15;14(10):20635-57. doi: 10.3390/ijms141020635. Int J Mol Sci. 2013. PMID: 24132151 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
