

Murine muscle cell models for Pompe disease and their use in studying therapeutic approaches
- PMID: 19167256
- PMCID: PMC2680079
- DOI: 10.1016/j.ymgme.2008.12.012
Murine muscle cell models for Pompe disease and their use in studying therapeutic approaches
Abstract
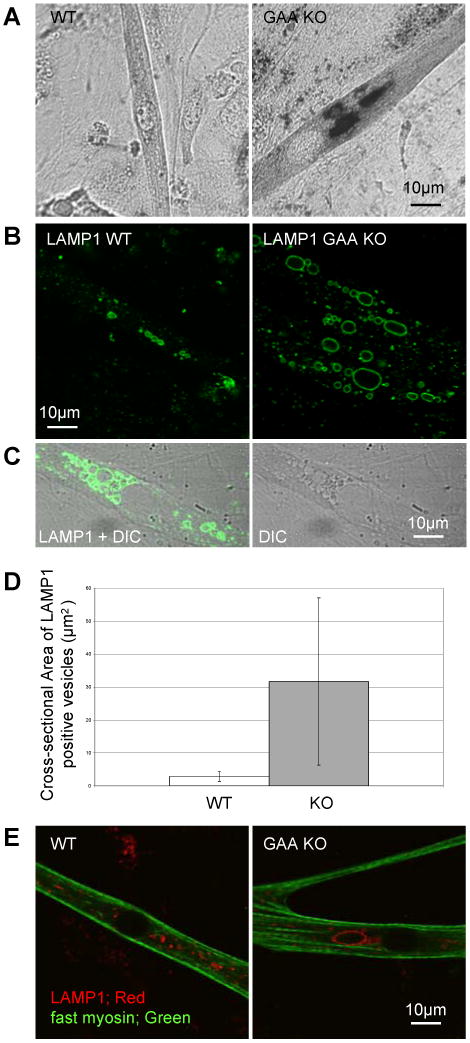
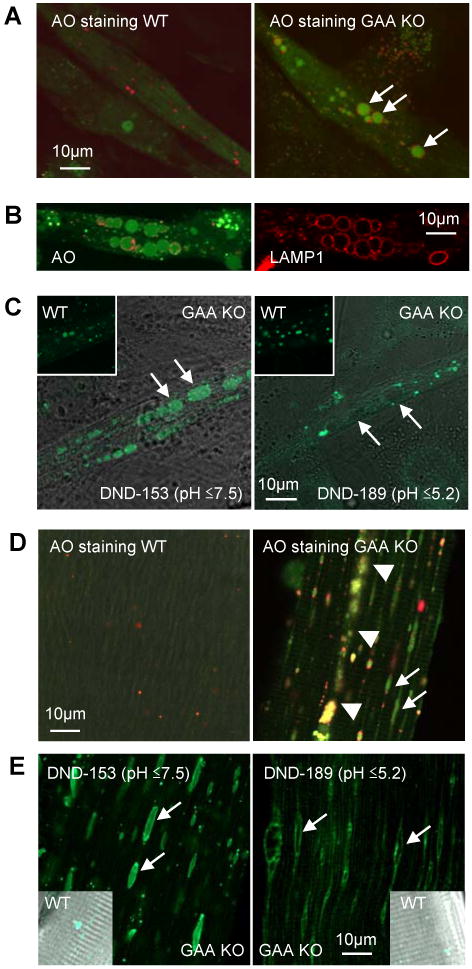
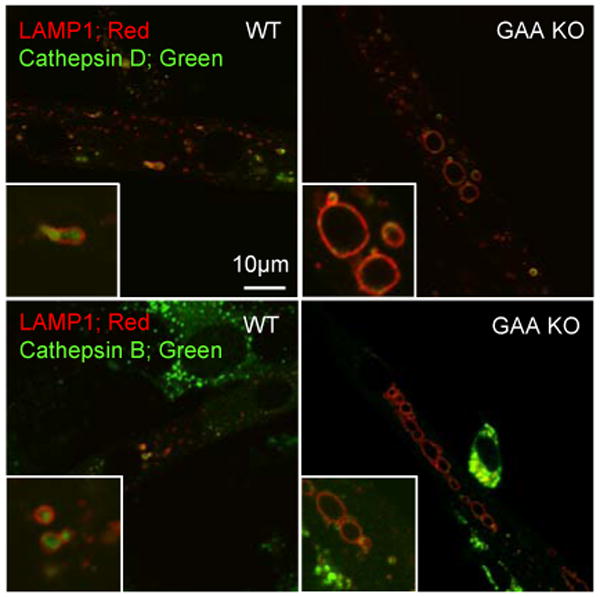
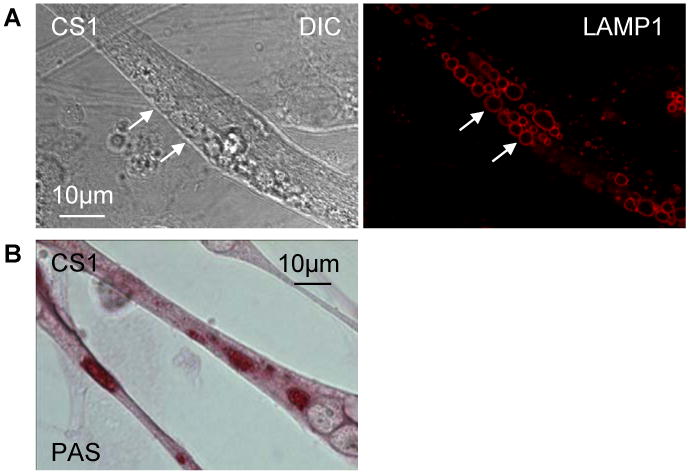
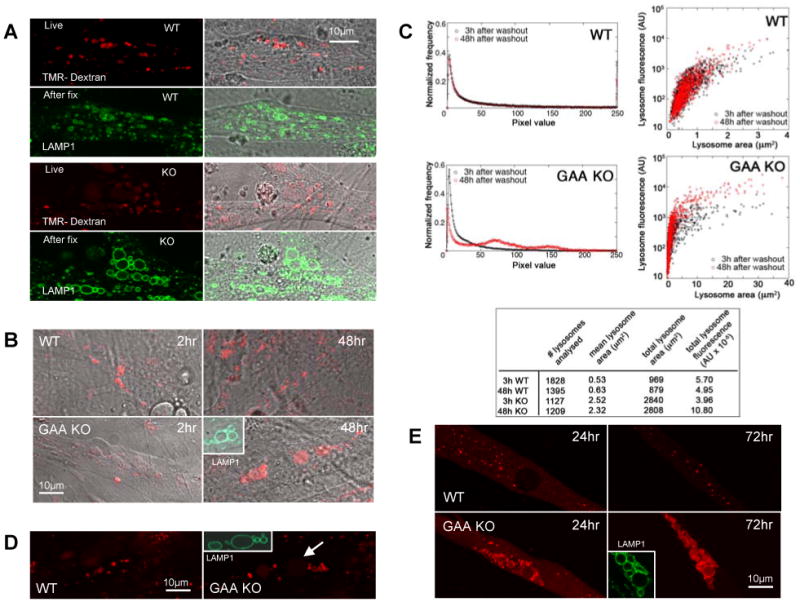
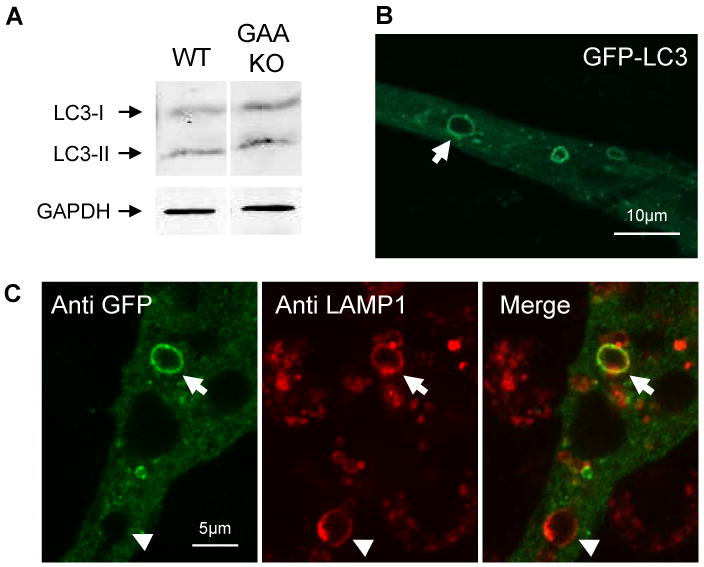
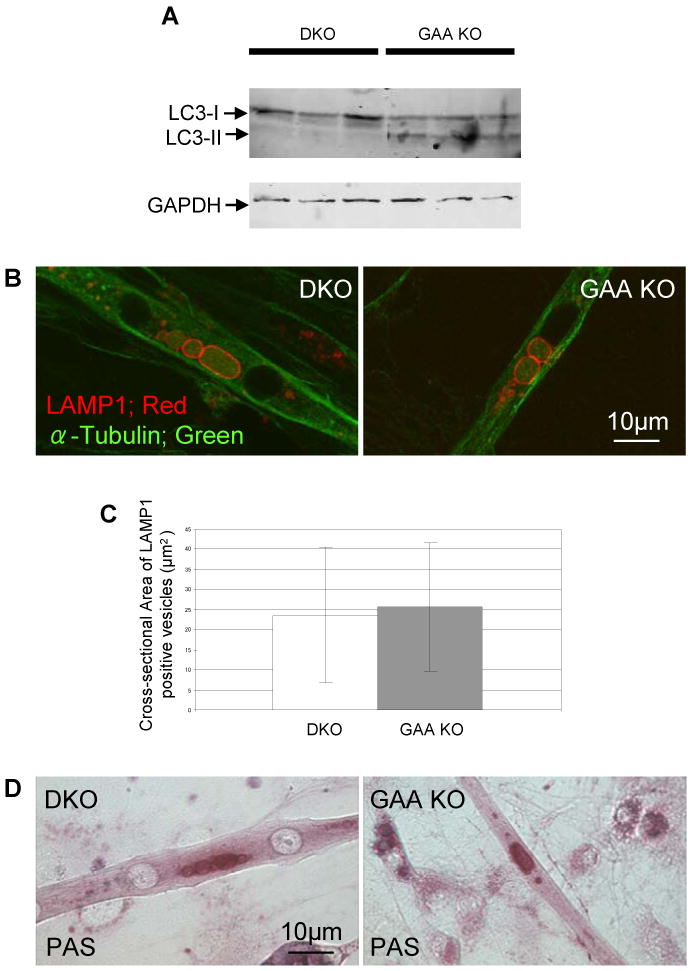
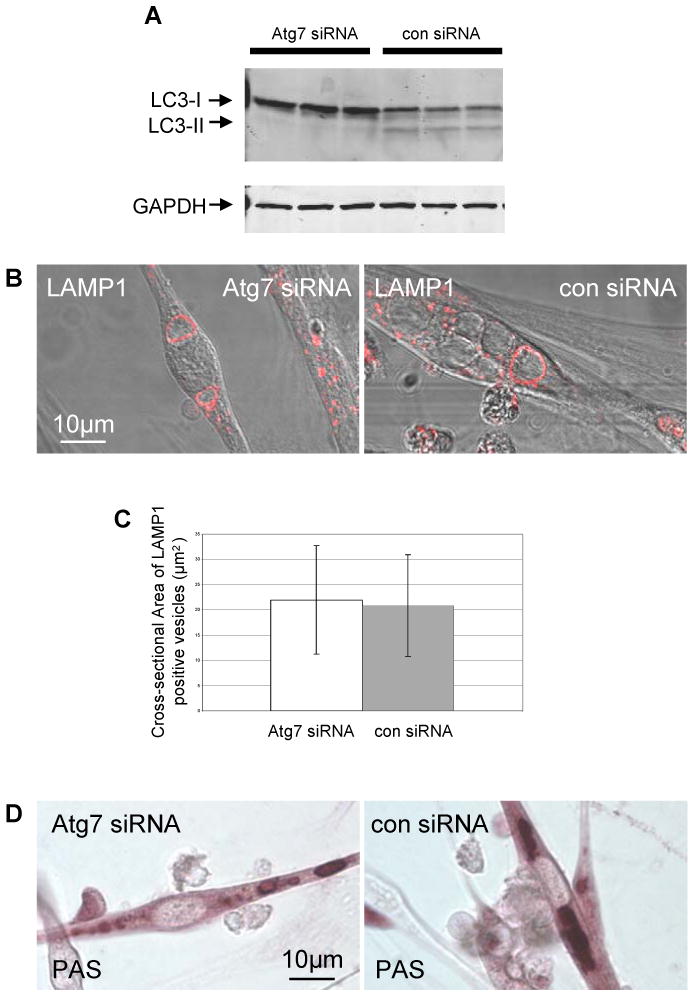
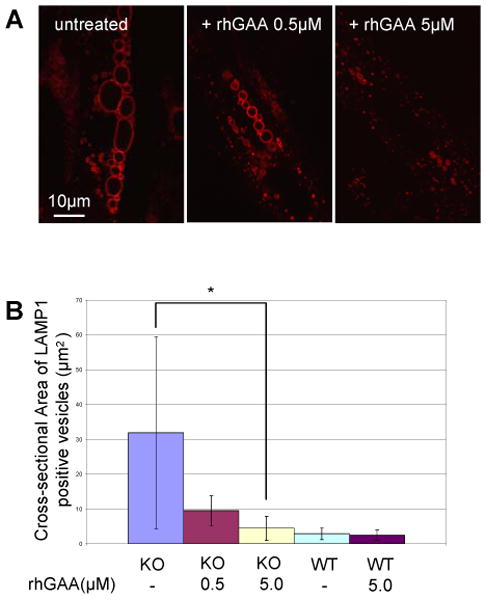
Lysosomes filled with glycogen are a major pathologic feature of Pompe disease, a fatal myopathy and cardiomyopathy caused by a deficiency of the glycogen-degrading lysosomal enzyme, acid alpha-glucosidase (GAA). To facilitate studies germane to this genetic disorder, we developed two in vitro Pompe models: myotubes derived from cultured primary myoblasts isolated from Pompe (GAA KO) mice, and myotubes derived from primary myoblasts of the same genotype that had been transduced with cyclin-dependent kinase 4 (CDK4). This latter model is endowed with extended proliferative capacity. Both models showed extremely large alkalinized, glycogen-filled lysosomes as well as impaired trafficking to lysosomes. Although both Pompe tissue culture models were derived from fast muscles and were fast myosin positive, they strongly resemble slow fibers in terms of their pathologic phenotype and their response to therapy with recombinant human GAA (rhGAA). Autophagic buildup, a hallmark of Pompe disease in fast muscle fibers, was absent, but basal autophagy was functional. To evaluate substrate deprivation as a strategy to prevent the accumulation of lysosomal glycogen, we knocked down Atg7, a gene essential for autophagosome formation, via siRNA, but we observed no effect on the extent of glycogen accumulation, thus confirming our recent observation in autophagy-deficient Pompe mice [N. Raben, V. Hill, L. Shea, S. Takikita, R. Baum, N. Mizushima, E. Ralston, P. Plotz, Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease, Hum. Mol. Genet. 17 (2008) 3897-3908] that macroautophagy is not the major route of glycogen transport to lysosomes. The in vitro Pompe models should be useful in addressing fundamental questions regarding the pathway of glycogen to the lysosomes and testing panels of small molecules that could affect glycogen biosynthesis or speed delivery of the replacement enzyme to affected lysosomes.
Figures
Similar articles
-
The values and limits of an in vitro model of Pompe disease: the best laid schemes o' mice an' men..Autophagy. 2009 Jul;5(5):729-31. doi: 10.4161/auto.5.5.8525. Autophagy. 2009. PMID: 19571661 Free PMC article.
-
Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder--murine Pompe disease.Autophagy. 2010 Nov;6(8):1078-89. doi: 10.4161/auto.6.8.13378. Autophagy. 2010. PMID: 20861693 Free PMC article.
-
Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease.Mol Ther. 2006 Dec;14(6):831-9. doi: 10.1016/j.ymthe.2006.08.009. Epub 2006 Sep 27. Mol Ther. 2006. PMID: 17008131 Free PMC article.
-
Autophagy in skeletal muscle: implications for Pompe disease.Int J Clin Pharmacol Ther. 2009;47 Suppl 1(Suppl 1):S42-7. doi: 10.5414/cpp47042. Int J Clin Pharmacol Ther. 2009. PMID: 20040311 Free PMC article. Review.
-
Autophagy and mitochondria in Pompe disease: nothing is so new as what has long been forgotten.Am J Med Genet C Semin Med Genet. 2012 Feb 15;160C(1):13-21. doi: 10.1002/ajmg.c.31317. Epub 2012 Jan 17. Am J Med Genet C Semin Med Genet. 2012. PMID: 22253254 Free PMC article. Review.
Cited by
-
Role of autophagy in glycogen breakdown and its relevance to chloroquine myopathy.PLoS Biol. 2013 Nov;11(11):e1001708. doi: 10.1371/journal.pbio.1001708. Epub 2013 Nov 12. PLoS Biol. 2013. PMID: 24265594 Free PMC article.
-
Micropatterned substrates with physiological stiffness promote cell maturation and Pompe disease phenotype in human induced pluripotent stem cell-derived skeletal myocytes.Biotechnol Bioeng. 2019 Sep;116(9):2377-2392. doi: 10.1002/bit.27075. Epub 2019 Jun 20. Biotechnol Bioeng. 2019. PMID: 31131875 Free PMC article.
-
Pompe disease results in a Golgi-based glycosylation deficit in human induced pluripotent stem cell-derived cardiomyocytes.J Biol Chem. 2015 Jan 30;290(5):3121-36. doi: 10.1074/jbc.M114.628628. Epub 2014 Dec 8. J Biol Chem. 2015. PMID: 25488666 Free PMC article.
-
Suppression of mTORC1 activation in acid-α-glucosidase-deficient cells and mice is ameliorated by leucine supplementation.Am J Physiol Regul Integr Comp Physiol. 2014 Nov 15;307(10):R1251-9. doi: 10.1152/ajpregu.00212.2014. Epub 2014 Sep 17. Am J Physiol Regul Integr Comp Physiol. 2014. PMID: 25231351 Free PMC article.
-
Biomedical Implications of Autophagy in Macromolecule Storage Disorders.Front Cell Dev Biol. 2019 Sep 6;7:179. doi: 10.3389/fcell.2019.00179. eCollection 2019. Front Cell Dev Biol. 2019. PMID: 31555645 Free PMC article. Review.
References
-
- Hirschhorn R, Reuser AJ. Glycogen storage disease type II; acid alpha-glucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited disease. McGraw-Hill; New York: 2001. pp. 3389–3420.
-
- Fukuda T, Roberts A, Plotz P, Raben N. Acid alpha-glucosidase deficiency (Pompe disease) Curr Neurol Neurosci Rep. 2007;7:71–77. - PubMed
-
- van der Ploeg A, Reuser A. Pompe's disease. Lancet. 2008;372:1342–1353. - PubMed
-
- Engel AG, Hirschhorn R, Huie ML. Acid Maltase Deficiency. In: Engel AG, Franzini-Armstrong C, editors. Myology. McGraw-Hill; New York: 2004. pp. 1559–1586.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
