

ATR and Chk1 suppress a caspase-3-dependent apoptotic response following DNA replication stress
- PMID: 19119425
- PMCID: PMC2607051
- DOI: 10.1371/journal.pgen.1000324
ATR and Chk1 suppress a caspase-3-dependent apoptotic response following DNA replication stress
Abstract
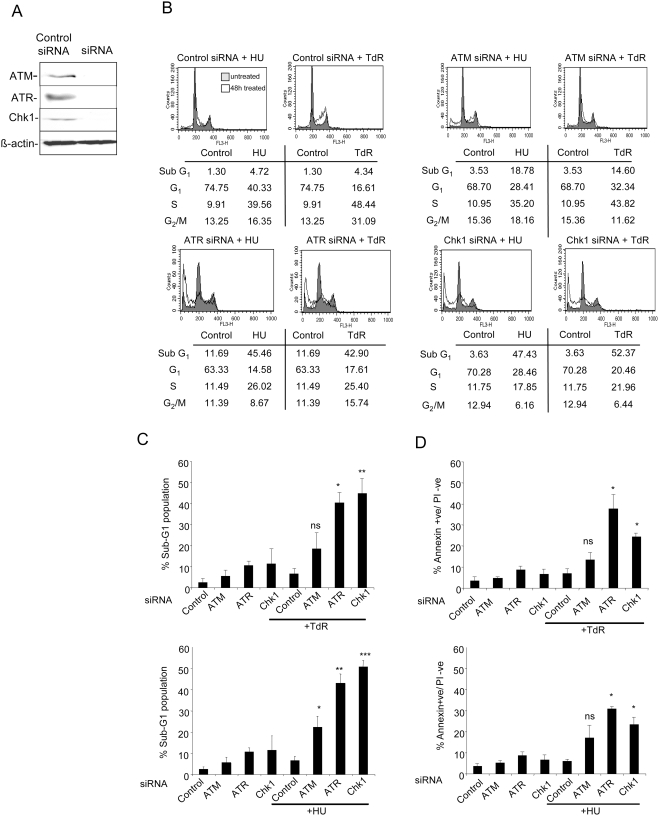
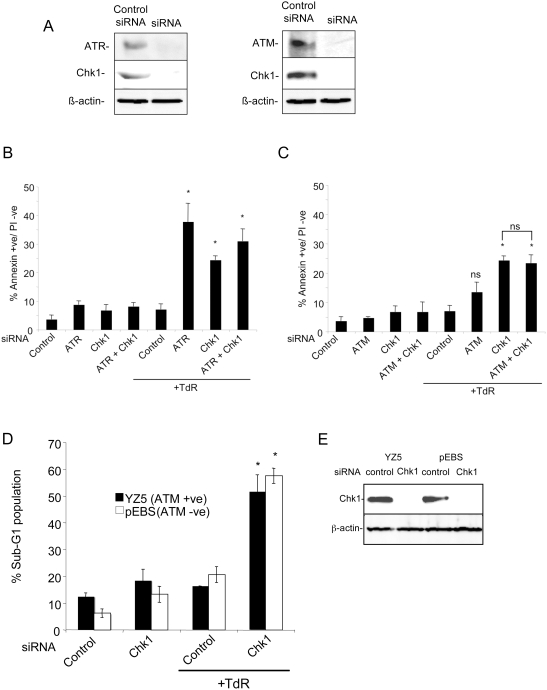
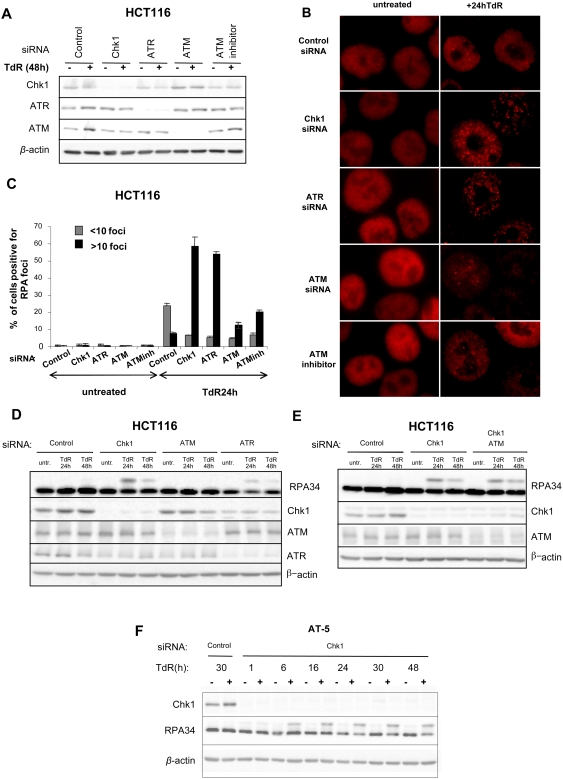
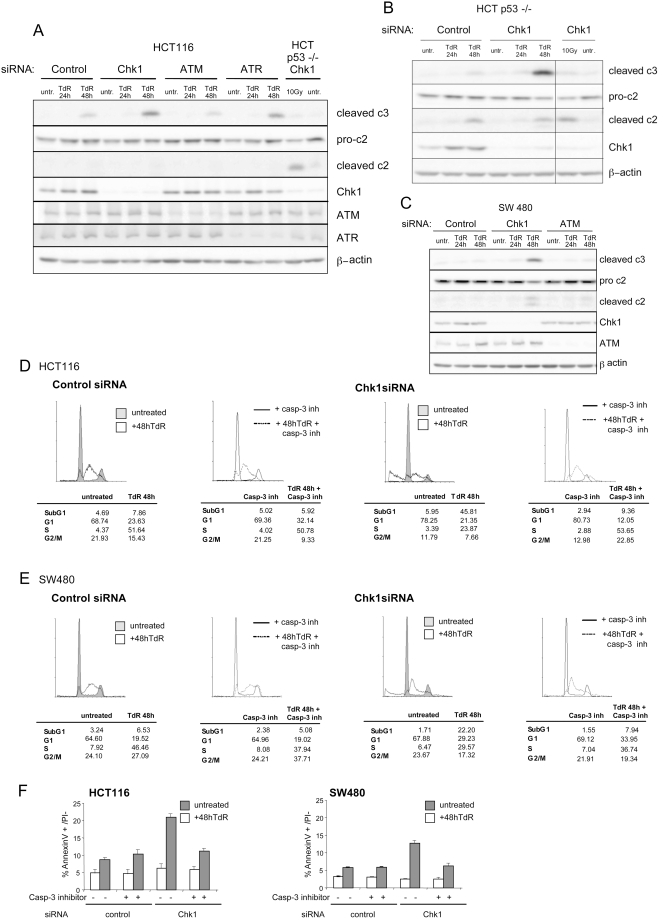
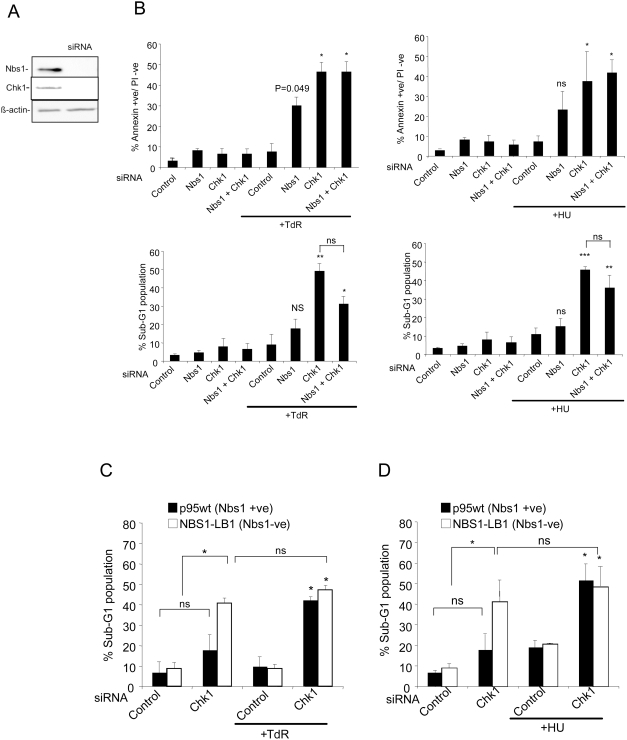
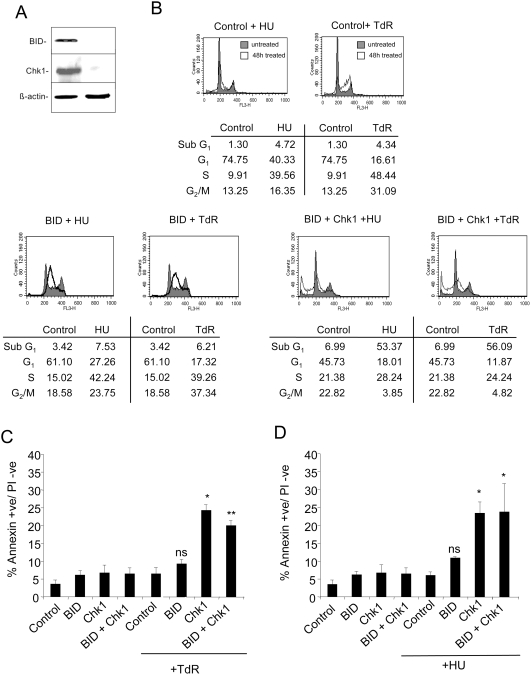
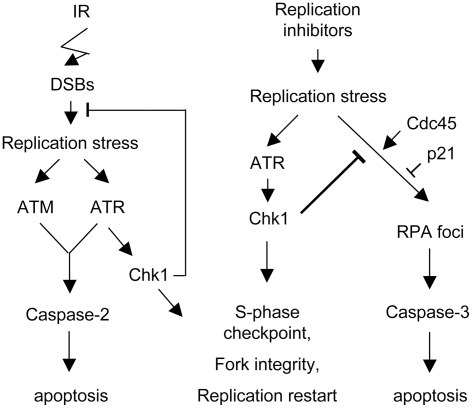
The related PIK-like kinases Ataxia-Telangiectasia Mutated (ATM) and ATM- and Rad3-related (ATR) play major roles in the regulation of cellular responses to DNA damage or replication stress. The pro-apoptotic role of ATM and p53 in response to ionizing radiation (IR) has been widely investigated. Much less is known about the control of apoptosis following DNA replication stress. Recent work indicates that Chk1, the downstream phosphorylation target of ATR, protects cells from apoptosis induced by DNA replication inhibitors as well as IR. The aim of the work reported here was to determine the roles of ATM- and ATR-protein kinase cascades in the control of apoptosis following replication stress and the relationship between Chk1-suppressed apoptotic pathways responding to replication stress or IR. ATM and ATR/Chk1 signalling pathways were manipulated using siRNA-mediated depletions or specific inhibitors in two tumour cell lines or fibroblasts derived from patients with inherited mutations. We show that depletion of ATM or its downstream phosphorylation targets, NBS1 and BID, has relatively little effect on apoptosis induced by DNA replication inhibitors, while ATR or Chk1 depletion strongly enhances cell death induced by such agents in all cells tested. Furthermore, early events occurring after the disruption of DNA replication (accumulation of RPA foci and RPA34 hyperphosphorylation) in ATR- or Chk1-depleted cells committed to apoptosis are not detected in ATM-depleted cells. Unlike the Chk1-suppressed pathway responding to IR, the replication stress-triggered apoptotic pathway did not require ATM and is characterized by activation of caspase 3 in both p53-proficient and -deficient cells. Taken together, our results show that the ATR-Chk1 signalling pathway plays a major role in the regulation of death in response to DNA replication stress and that the Chk1-suppressed pathway protecting cells from replication stress is clearly distinguishable from that protecting cells from IR.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation.J Biol Chem. 2003 Apr 25;278(17):14806-11. doi: 10.1074/jbc.M210862200. Epub 2003 Feb 14. J Biol Chem. 2003. PMID: 12588868
-
Dissecting cellular responses to irradiation via targeted disruptions of the ATM-CHK1-PP2A circuit.Cell Cycle. 2013 Apr 1;12(7):1105-18. doi: 10.4161/cc.24127. Epub 2013 Mar 5. Cell Cycle. 2013. PMID: 23462183 Free PMC article.
-
Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selective target specificities in response to different forms of DNA damage.J Biol Chem. 2005 Jan 14;280(2):1186-92. doi: 10.1074/jbc.M410873200. Epub 2004 Nov 8. J Biol Chem. 2005. PMID: 15533933
-
The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer.Adv Cancer Res. 2010;108:73-112. doi: 10.1016/B978-0-12-380888-2.00003-0. Adv Cancer Res. 2010. PMID: 21034966 Review.
-
Regulation of ATR-CHK1 signaling by ubiquitination of CLASPIN.Biochem Soc Trans. 2022 Oct 31;50(5):1471-1480. doi: 10.1042/BST20220729. Biochem Soc Trans. 2022. PMID: 36196914 Review.
Cited by
-
Checkpoint kinase 1 in DNA damage response and cell cycle regulation.Cell Mol Life Sci. 2013 Nov;70(21):4009-21. doi: 10.1007/s00018-013-1307-3. Epub 2013 Mar 19. Cell Mol Life Sci. 2013. PMID: 23508805 Free PMC article. Review.
-
A New Way to Treat Brain Tumors: Targeting Proteins Coded by Microcephaly Genes?: Brain tumors and microcephaly arise from opposing derangements regulating progenitor growth. Drivers of microcephaly could be attractive brain tumor targets.Bioessays. 2018 May;40(5):e1700243. doi: 10.1002/bies.201700243. Epub 2018 Mar 26. Bioessays. 2018. PMID: 29577351 Free PMC article. Review.
-
Impaired tissue growth is mediated by checkpoint kinase 1 (CHK1) in the integrated stress response.J Cell Sci. 2010 Sep 1;123(Pt 17):2892-900. doi: 10.1242/jcs.070078. Epub 2010 Aug 3. J Cell Sci. 2010. PMID: 20682638 Free PMC article.
-
Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation.Cell Death Dis. 2012 Dec 6;3(12):e441. doi: 10.1038/cddis.2012.181. Cell Death Dis. 2012. PMID: 23222511 Free PMC article.
-
RNF4 and USP7 cooperate in ubiquitin-regulated steps of DNA replication.Open Biol. 2023 Aug;13(8):230068. doi: 10.1098/rsob.230068. Epub 2023 Aug 23. Open Biol. 2023. PMID: 37607592 Free PMC article.
References
-
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168. - PubMed
-
- Falck J, Malland N, Syljuasen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–847. - PubMed
-
- Falck J, Petrini JH, Williams BR, Lukas J, Bartek J. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet. 2002;30:290–294. - PubMed
-
- Lim DS, Kim ST, Xu B, Maser RS, Lin J, et al. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000;404:613–617. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
