

The FMR1 gene and fragile X-associated tremor/ataxia syndrome
- PMID: 19105204
- PMCID: PMC4320942
- DOI: 10.1002/ajmg.b.30910
The FMR1 gene and fragile X-associated tremor/ataxia syndrome
Abstract
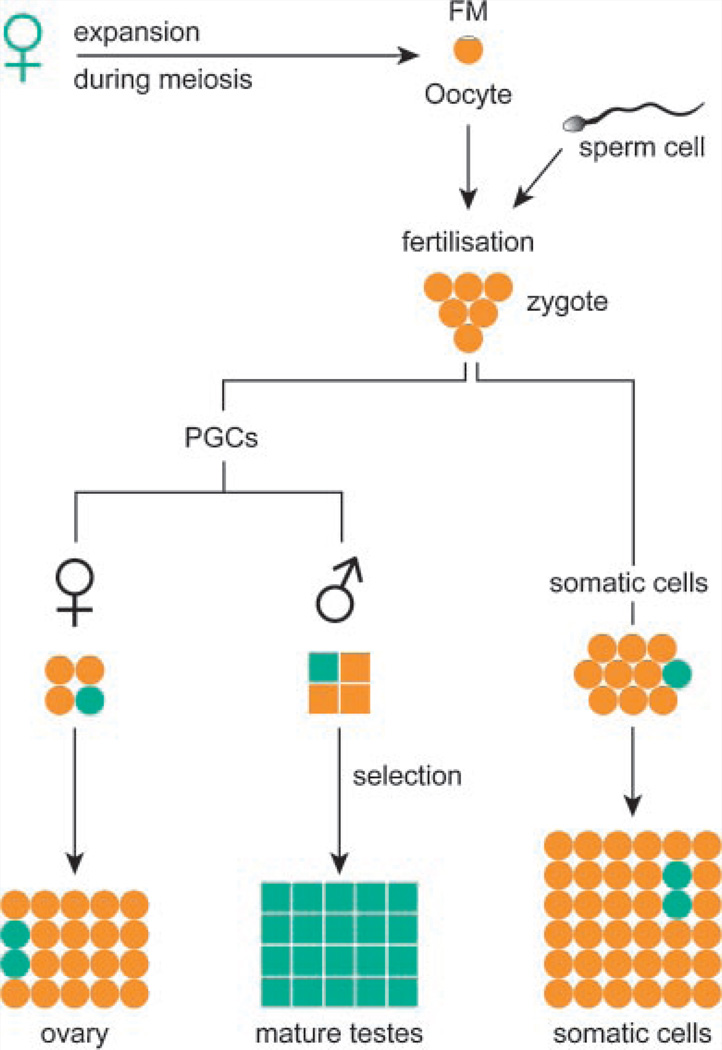
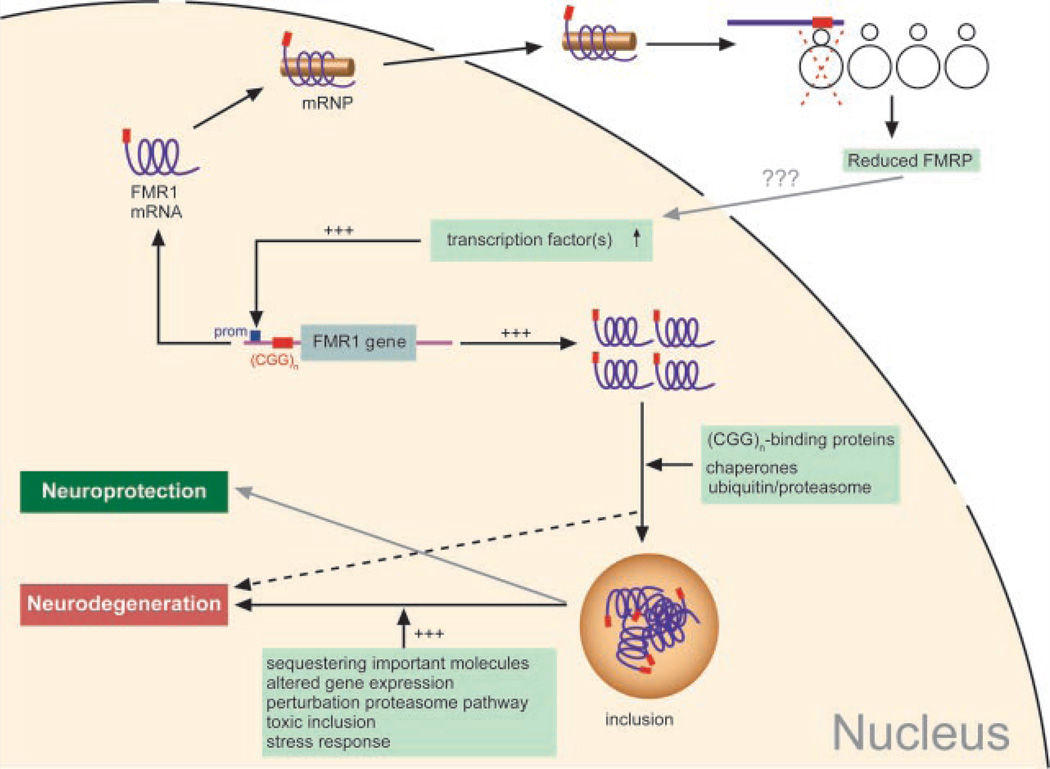
The CGG-repeat present in the 5'UTR of the FMR1 gene is unstable upon transmission to the next generation. The repeat is up to 55 CGGs long in the normal population. In fragile X patients, a repeat length exceeding 200 CGGs (full mutation: FM) generally leads to methylation of the repeat and the promoter region, which is accompanied by silencing of the FMR1 gene. The gene product FMRP is involved in regulation of transport and translation of certain mRNA in the dendrite, thereby affecting synaptic plasticity. This is central to learning and memory processes. The absence of FMRP seen in FM is the cause of the mental retardation seen in fragile X patients. The premutation (PM) is defined as 55-200 CGGs. Female PM carriers are at risk of developing primary ovarian insufficiency. Recently it was discovered that elderly PM carriers might develop a progressive neurodegenerative disorder called fragile X-associated tremor/ataxia syndrome. Although arising from the mutations in the same gene, distinct mechanisms lead to fragile X syndrome (absence of FMRP) and FXTAS (toxic RNA gain of function). The pathogenic mechanisms thought to underlie these disorders are discussed, with a specific emphasis on FXTAS. This review gives insight on the implications of all possible repeat length categories seen in fragile X families.
(c) 2008 Wiley-Liss, Inc.
Figures


Similar articles
-
CGG repeat in the FMR1 gene: size matters.Clin Genet. 2011 Sep;80(3):214-25. doi: 10.1111/j.1399-0004.2011.01723.x. Epub 2011 Jun 30. Clin Genet. 2011. PMID: 21651511 Free PMC article. Review.
-
Presence of inclusions positive for polyglycine containing protein, FMRpolyG, indicates that repeat-associated non-AUG translation plays a role in fragile X-associated primary ovarian insufficiency.Hum Reprod. 2016 Jan;31(1):158-68. doi: 10.1093/humrep/dev280. Epub 2015 Nov 3. Hum Reprod. 2016. PMID: 26537920 Free PMC article.
-
Altered hypothalamus-pituitary-adrenal gland axis regulation in the expanded CGG-repeat mouse model for fragile X-associated tremor/ataxia syndrome.Psychoneuroendocrinology. 2008 Jul;33(6):863-73. doi: 10.1016/j.psyneuen.2008.03.011. Epub 2008 May 12. Psychoneuroendocrinology. 2008. PMID: 18472227 Free PMC article.
-
Unstable mutations in the FMR1 gene and the phenotypes.Adv Exp Med Biol. 2012;769:78-114. doi: 10.1007/978-1-4614-5434-2_6. Adv Exp Med Biol. 2012. PMID: 23560306 Free PMC article. Review.
-
[FMR1 PREMUTATION CARRIERS - ARE THEY REALLY ASYMPTOMATIC?].Harefuah. 2018 Apr;157(4):241-244. Harefuah. 2018. PMID: 29688643 Review. Hebrew.
Cited by
-
Neuropathology of FMR1-premutation carriers presenting with dementia and neuropsychiatric symptoms.Brain Commun. 2021 Jan 27;3(1):fcab007. doi: 10.1093/braincomms/fcab007. eCollection 2021. Brain Commun. 2021. PMID: 33709078 Free PMC article.
-
Genetic and Environmental Contributions to Autism Spectrum Disorder Through Mechanistic Target of Rapamycin.Biol Psychiatry Glob Open Sci. 2021 Sep 1;2(2):95-105. doi: 10.1016/j.bpsgos.2021.08.005. eCollection 2022 Apr. Biol Psychiatry Glob Open Sci. 2021. PMID: 36325164 Free PMC article. Review.
-
mTOR, a Potential Target to Treat Autism Spectrum Disorder.CNS Neurol Disord Drug Targets. 2016;15(5):533-43. doi: 10.2174/1871527315666160413120638. CNS Neurol Disord Drug Targets. 2016. PMID: 27071790 Free PMC article. Review.
-
Population-based estimates of the prevalence of FMR1 expansion mutations in women with early menopause and primary ovarian insufficiency.Genet Med. 2014 Jan;16(1):19-24. doi: 10.1038/gim.2013.64. Epub 2013 May 23. Genet Med. 2014. PMID: 23703681 Free PMC article.
-
Association of skewed X-chromosome inactivation with FMR1 CGG repeat length and anti-Mullerian hormone levels: a cohort study.Reprod Biol Endocrinol. 2017 Apr 28;15(1):34. doi: 10.1186/s12958-017-0250-9. Reprod Biol Endocrinol. 2017. PMID: 28454580 Free PMC article.
References
-
- Abitbol M, Menini C, Delezoide AL, Rhyner T, Vekemans M, Mallet J. Nucleus basalis magnocellularis and hippocampus are the major sites of FMR-1 expression in the human fetal brain. Nat Genet. 1993;4:147–153. - PubMed
-
- Agulhon C, Blanchet P, Kobetz A, Marchant D, Faucon N, Sarda P, Moraine C, Sittler A, Biancalana V, Malafosse A, et al. Expression of FMR1, FXR1, and FXR2 genes in human prenatal tissues. J Neuropathol Exp Neurol. 1999;58:867–880. - PubMed
-
- Allen EG, He W, Yadav-Shah M, Sherman SL. A study of the distributional characteristics ofFMR1transcript levels in 238 individuals. Hum Genet. 2004;114:439–447. - PubMed
-
- Allen EG, Sullivan AK, Marcus M, Small C, Dominguez C, Epstein MP, Charen K, He W, Taylor KC, Sherman SL. Examination of reproductive aging milestones among women who carry the FMR1 premutation. Hum Reprod. 2007;22:2142–2152. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
