

p120 catenin induces opposing effects on tumor cell growth depending on E-cadherin expression
- PMID: 19015320
- PMCID: PMC2582886
- DOI: 10.1083/jcb.200805113
p120 catenin induces opposing effects on tumor cell growth depending on E-cadherin expression
Abstract
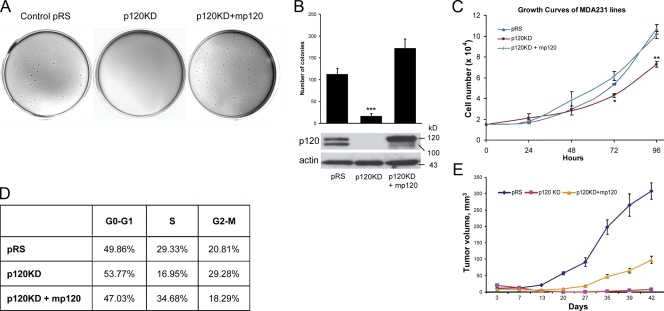
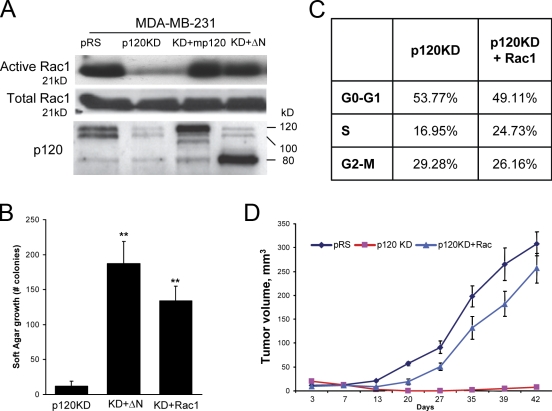
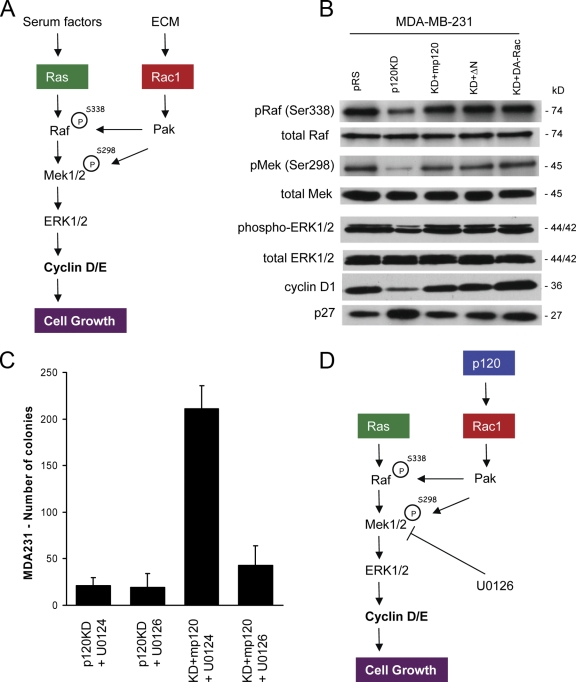
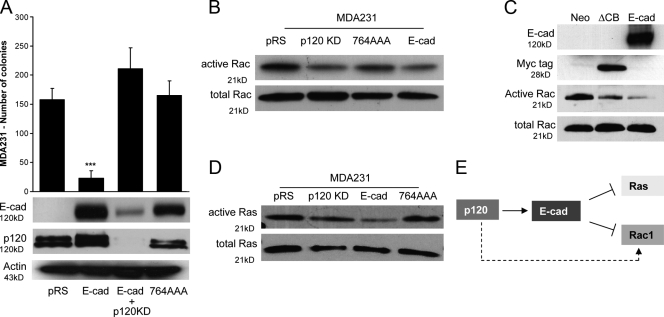
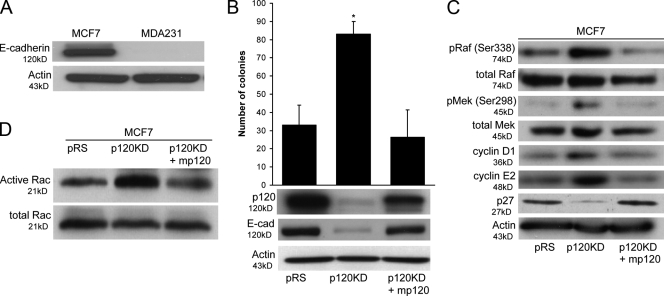
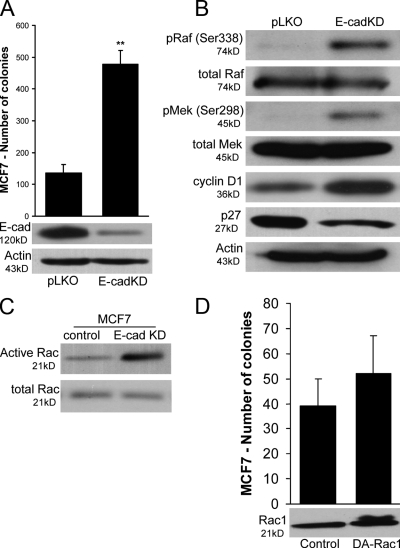
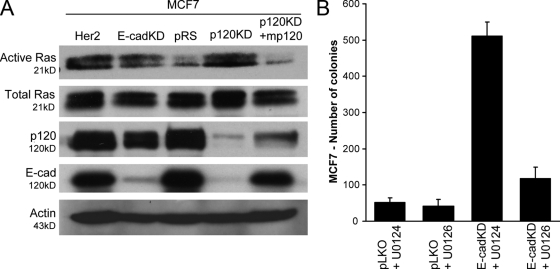
p120 catenin regulates the activity of the Rho family guanosine triphosphatases (including RhoA and Rac1) in an adhesion-dependent manner. Through this action, p120 promotes a sessile cellular phenotype when associated with epithelial cadherin (E-cadherin) or a motile phenotype when associated with mesenchymal cadherins. In this study, we show that p120 also exerts significant and diametrically opposing effects on tumor cell growth depending on E-cadherin expression. Endogenous p120 acts to stabilize E-cadherin complexes and to actively promote the tumor-suppressive function of E-cadherin, potently inhibiting Ras activation. Upon E-cadherin loss during tumor progression, the negative regulation of Ras is relieved; under these conditions, endogenous p120 promotes transformed cell growth both in vitro and in vivo by activating a Rac1-mitogen-activated protein kinase signaling pathway normally activated by the adhesion of cells to the extracellular matrix. These data indicate that both E-cadherin and p120 are important regulators of tumor cell growth and imply roles for both proteins in chemoresistance and targeted therapeutics.
Figures

Similar articles
-
p120 catenin is essential for mesenchymal cadherin-mediated regulation of cell motility and invasiveness.J Cell Biol. 2006 Sep 25;174(7):1087-96. doi: 10.1083/jcb.200605022. Epub 2006 Sep 18. J Cell Biol. 2006. PMID: 16982802 Free PMC article.
-
A core function for p120-catenin in cadherin turnover.J Cell Biol. 2003 Nov 10;163(3):525-34. doi: 10.1083/jcb.200307111. J Cell Biol. 2003. PMID: 14610055 Free PMC article.
-
Inhibition of RhoA by p120 catenin.Nat Cell Biol. 2000 Sep;2(9):637-44. doi: 10.1038/35023588. Nat Cell Biol. 2000. PMID: 10980705
-
p120-ctn: A nexus for contextual signaling via Rho GTPases.Biochim Biophys Acta. 2007 Jan;1773(1):34-46. doi: 10.1016/j.bbamcr.2006.08.040. Epub 2006 Sep 1. Biochim Biophys Acta. 2007. PMID: 17028013 Review.
-
Emerging roles for p120-catenin in cell adhesion and cancer.Oncogene. 2004 Oct 18;23(48):7947-56. doi: 10.1038/sj.onc.1208161. Oncogene. 2004. PMID: 15489912 Review.
Cited by
-
Role of Cadherins in Cancer-A Review.Int J Mol Sci. 2020 Oct 15;21(20):7624. doi: 10.3390/ijms21207624. Int J Mol Sci. 2020. PMID: 33076339 Free PMC article. Review.
-
Phosphorylation of VE-cadherin controls endothelial phenotypes via p120-catenin coupling and Rac1 activation.Am J Physiol Heart Circ Physiol. 2011 Jan;300(1):H162-72. doi: 10.1152/ajpheart.00650.2010. Epub 2010 Oct 29. Am J Physiol Heart Circ Physiol. 2011. PMID: 21037229 Free PMC article.
-
Cytosolic p120-catenin regulates growth of metastatic lobular carcinoma through Rock1-mediated anoikis resistance.J Clin Invest. 2011 Aug;121(8):3176-88. doi: 10.1172/JCI41695. Epub 2011 Jul 11. J Clin Invest. 2011. PMID: 21747168 Free PMC article.
-
Cadherins and catenins in cancer: connecting cancer pathways and tumor microenvironment.Front Cell Dev Biol. 2023 May 15;11:1137013. doi: 10.3389/fcell.2023.1137013. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 37255594 Free PMC article. Review.
-
Prolonged overexpression of Wnt10b induces epidermal keratinocyte transformation through activating EGF pathway.Histochem Cell Biol. 2015 Sep;144(3):209-21. doi: 10.1007/s00418-015-1330-6. Epub 2015 May 21. Histochem Cell Biol. 2015. PMID: 25995040 Free PMC article.
References
-
- Anastasiadis, P.Z. 2007. p120-ctn: a nexus for contextual signaling via Rho GTPases. Biochim. Biophys. Acta. 1773:34–46. - PubMed
-
- Anastasiadis, P.Z., S.Y. Moon, M.A. Thoreson, D.J. Mariner, H.C. Crawford, Y. Zheng, and A.B. Reynolds. 2000. Inhibition of RhoA by p120 catenin. Nat. Cell Biol. 2:637–644. - PubMed
-
- Beeser, A., Z.M. Jaffer, C. Hofmann, and J. Chernoff. 2005. Role of group A p21-activated kinases in activation of extracellular-regulated kinase by growth factors. J. Biol. Chem. 280:36609–36615. - PubMed
-
- Betson, M., E. Lozano, J. Zhang, and V.M. Braga. 2002. Rac activation upon cell-cell contact formation is dependent on signaling from the epidermal growth factor receptor. J. Biol. Chem. 277:36962–36969. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
