

Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity
- PMID: 18957222
- PMCID: PMC2634746
- DOI: 10.1016/j.neuron.2008.08.012
Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity
Abstract
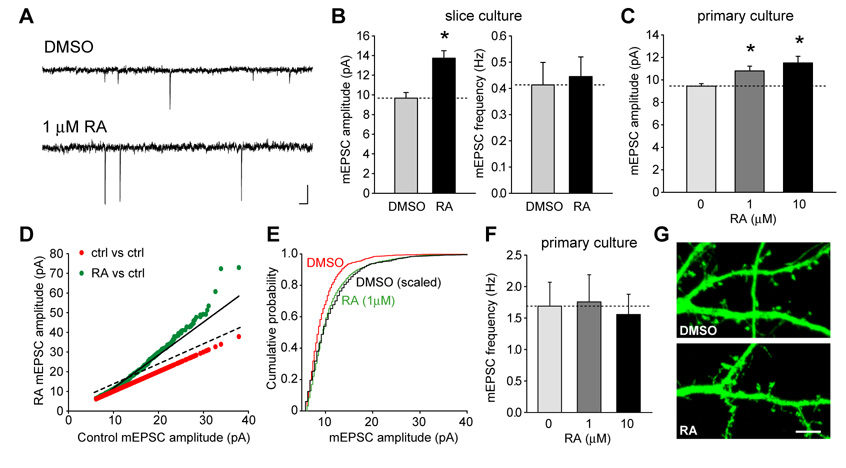
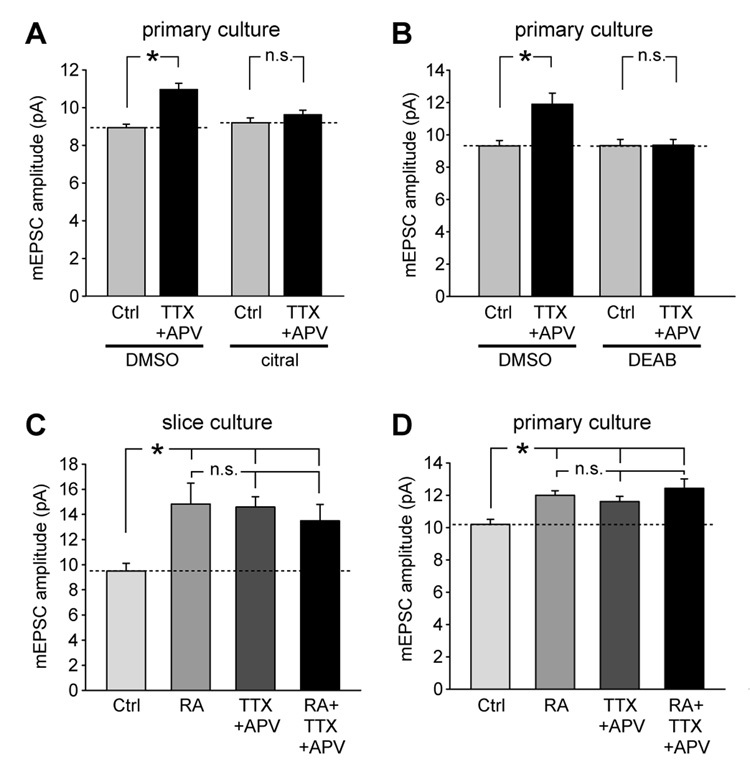
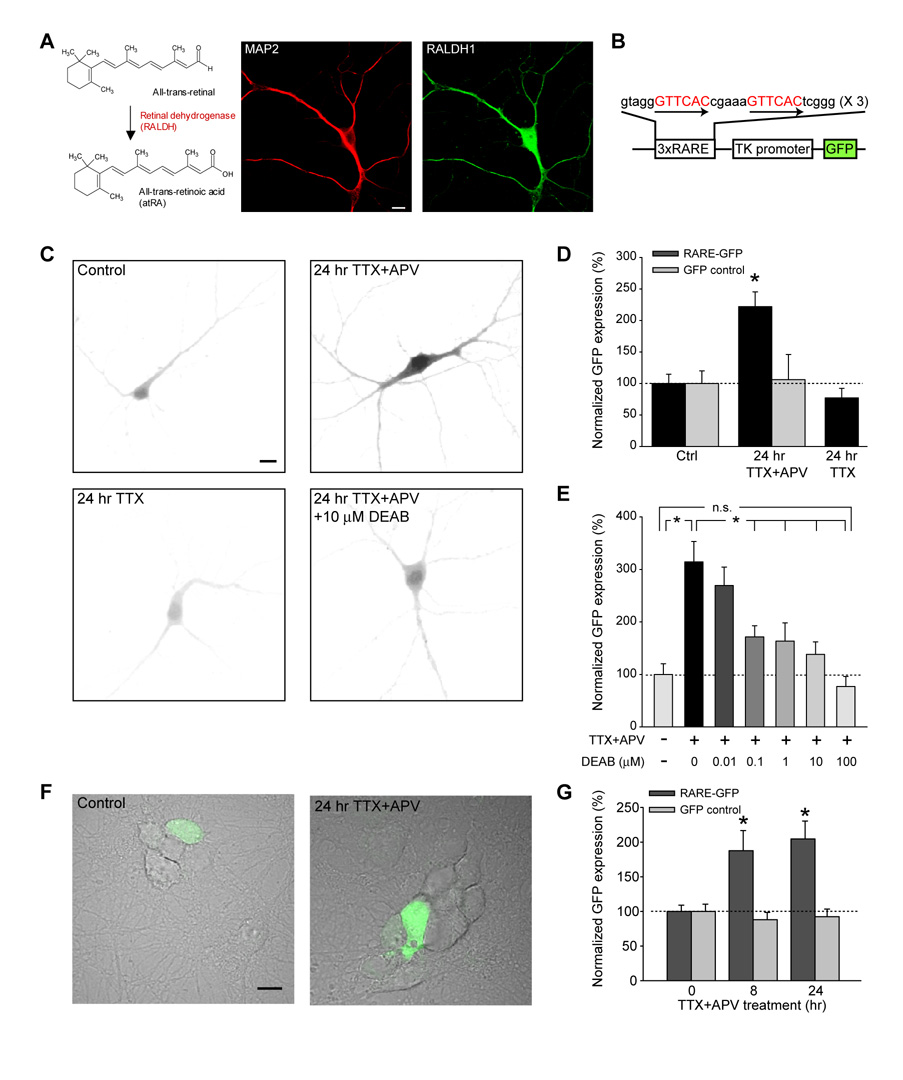
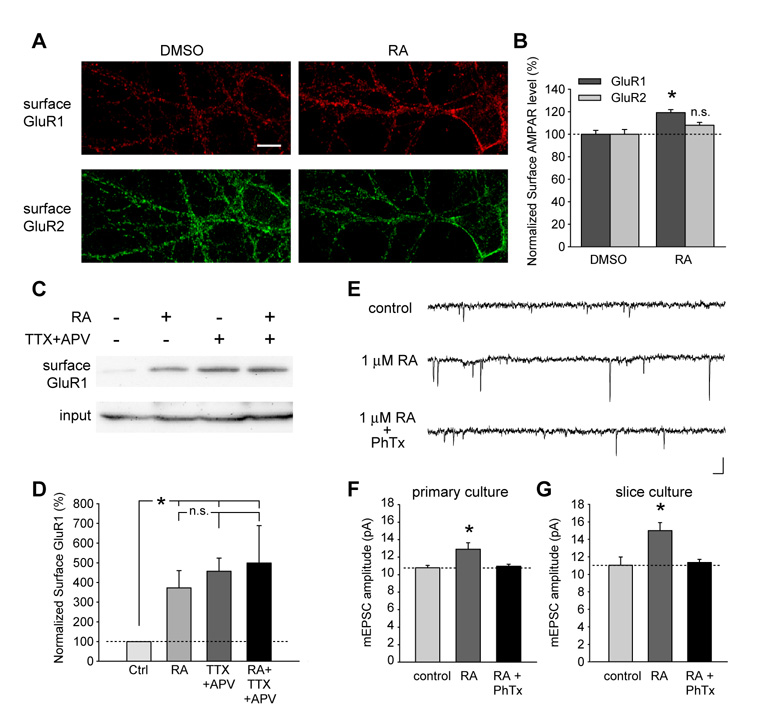
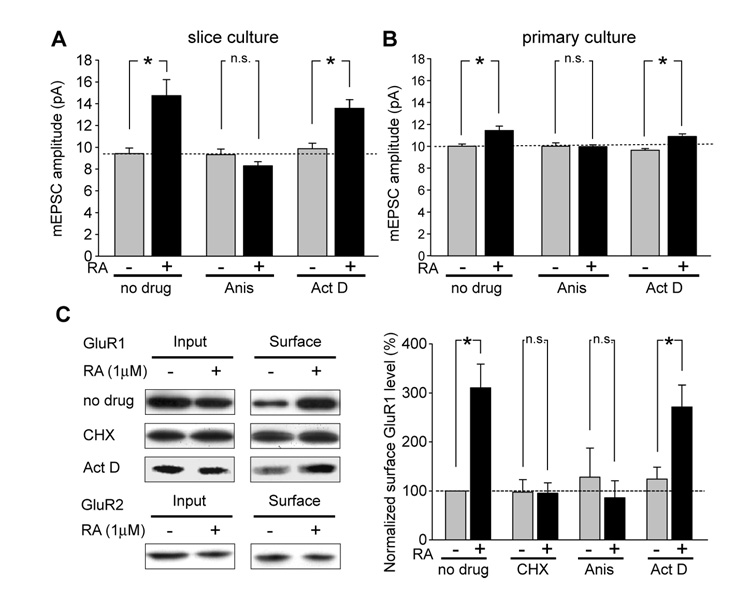
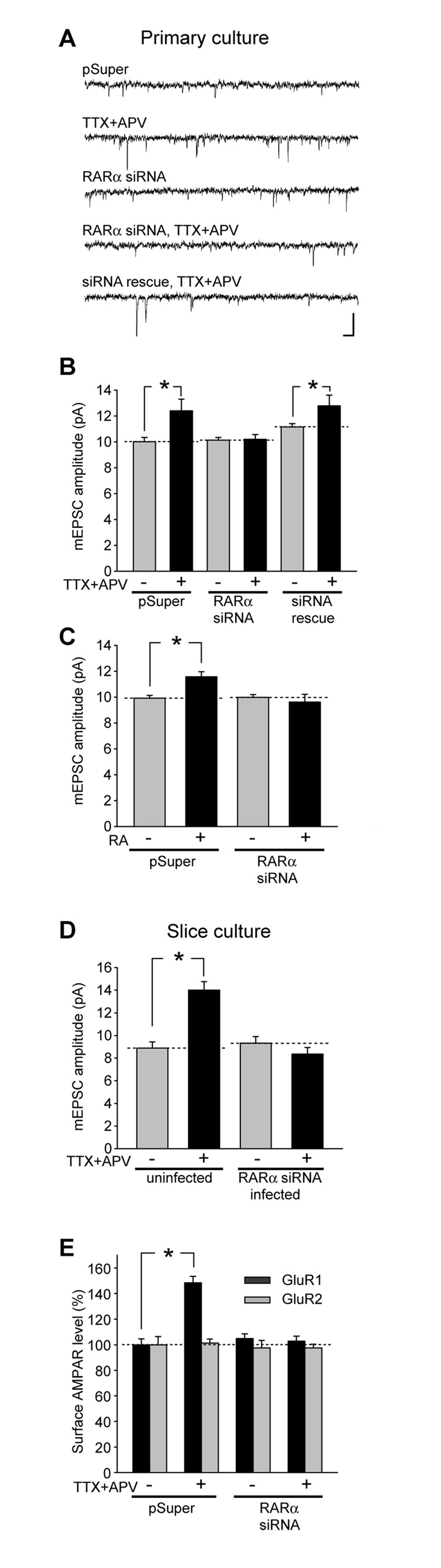
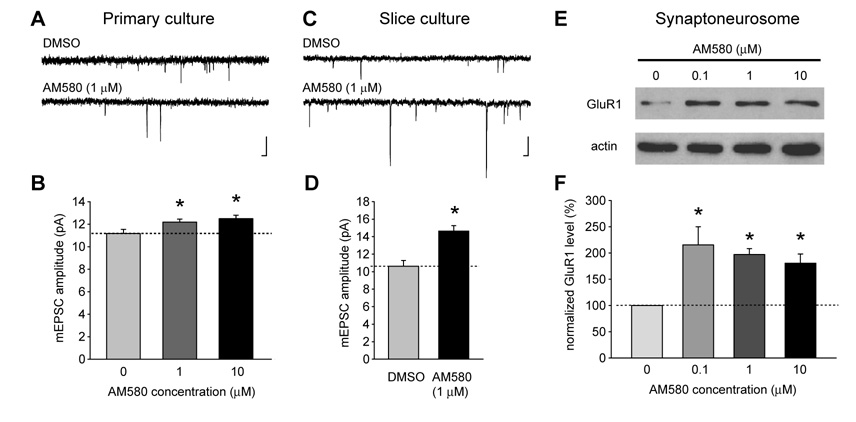
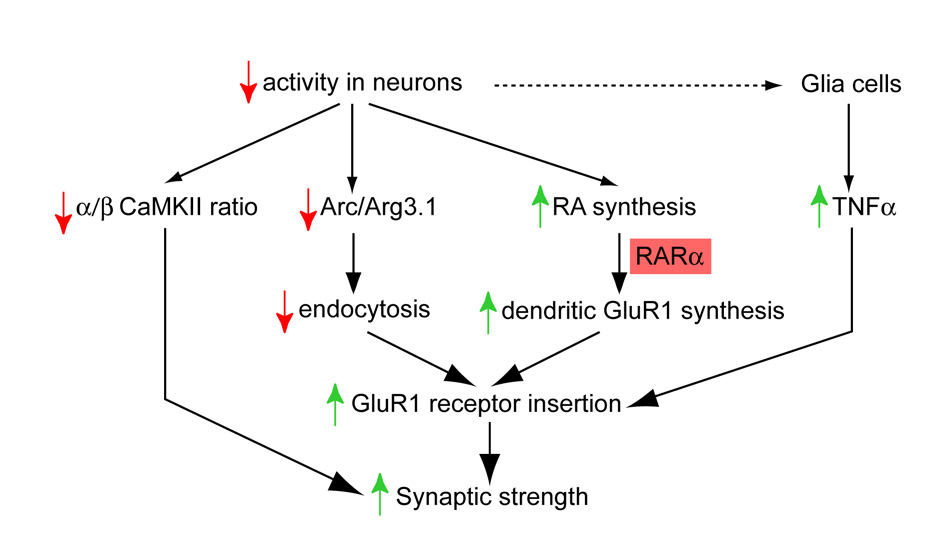
Normal brain function requires that the overall synaptic activity in neural circuits be kept constant. Long-term alterations of neural activity lead to homeostatic regulation of synaptic strength by a process known as synaptic scaling. The molecular mechanisms underlying synaptic scaling are largely unknown. Here, we report that all-trans retinoic acid (RA), a well-known developmental morphogen, unexpectedly mediates synaptic scaling in response to activity blockade. We show that activity blockade increases RA synthesis in neurons and that acute RA treatment enhances synaptic transmission. The RA-induced increase in synaptic strength is occluded by activity blockade-induced synaptic scaling. Suppression of RA synthesis prevents synaptic scaling. This form of RA signaling operates via a translation-dependent but transcription-independent mechanism, causes an upregulation of postsynaptic glutamate receptor levels, and requires RARalpha receptors. Together, our data suggest that RA functions in homeostatic plasticity as a signaling molecule that increases synaptic strength by a protein synthesis-dependent mechanism.
Figures

Comment in
-
A role for retinoic acid in homeostatic plasticity.Neuron. 2008 Oct 23;60(2):192-4. doi: 10.1016/j.neuron.2008.10.003. Neuron. 2008. PMID: 18957211 Free PMC article.
Similar articles
-
Synaptic retinoic acid receptor signaling mediates mTOR-dependent metaplasticity that controls hippocampal learning.Proc Natl Acad Sci U S A. 2019 Apr 2;116(14):7113-7122. doi: 10.1073/pnas.1820690116. Epub 2019 Feb 19. Proc Natl Acad Sci U S A. 2019. PMID: 30782829 Free PMC article.
-
Retinoic acid regulates RARalpha-mediated control of translation in dendritic RNA granules during homeostatic synaptic plasticity.Proc Natl Acad Sci U S A. 2008 Oct 14;105(41):16015-20. doi: 10.1073/pnas.0804801105. Epub 2008 Oct 7. Proc Natl Acad Sci U S A. 2008. PMID: 18840692 Free PMC article.
-
Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid.J Neurosci. 2010 Dec 15;30(50):16910-21. doi: 10.1523/JNEUROSCI.3660-10.2010. J Neurosci. 2010. PMID: 21159962 Free PMC article.
-
Synaptic retinoic acid signaling and homeostatic synaptic plasticity.Neuropharmacology. 2014 Mar;78:3-12. doi: 10.1016/j.neuropharm.2012.12.004. Epub 2012 Dec 25. Neuropharmacology. 2014. PMID: 23270606 Free PMC article. Review.
-
Insufficient support for retinoic acid receptor control of synaptic plasticity through a non-genomic mechanism.Front Neuroendocrinol. 2023 Oct;71:101099. doi: 10.1016/j.yfrne.2023.101099. Epub 2023 Aug 28. Front Neuroendocrinol. 2023. PMID: 37647946 Free PMC article. Review.
Cited by
-
miR-329- and miR-495-mediated Prr7 down-regulation is required for homeostatic synaptic depression in rat hippocampal neurons.Life Sci Alliance. 2022 Sep 23;5(12):e202201520. doi: 10.26508/lsa.202201520. Life Sci Alliance. 2022. PMID: 36150742 Free PMC article.
-
In vivo synaptic scaling is mediated by GluA2-lacking AMPA receptors in the embryonic spinal cord.J Neurosci. 2013 Apr 17;33(16):6791-9. doi: 10.1523/JNEUROSCI.4025-12.2013. J Neurosci. 2013. PMID: 23595738 Free PMC article.
-
Wherefore art thou, homeo(stasis)? Functional diversity in homeostatic synaptic plasticity.Neural Plast. 2012;2012:718203. doi: 10.1155/2012/718203. Epub 2012 May 17. Neural Plast. 2012. PMID: 22685679 Free PMC article. Review.
-
Genome-wide effects of social status on DNA methylation in the brain of a cichlid fish, Astatotilapia burtoni.BMC Genomics. 2019 Sep 11;20(1):699. doi: 10.1186/s12864-019-6047-9. BMC Genomics. 2019. PMID: 31506062 Free PMC article.
-
Cytochrome P450 26A1 Contributes to the Maintenance of Neuropathic Pain.Neurosci Bull. 2024 Mar;40(3):293-309. doi: 10.1007/s12264-023-01101-1. Epub 2023 Aug 28. Neurosci Bull. 2024. PMID: 37639183 Free PMC article.
References
-
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. - PubMed
-
- Blomhoff R, Green MH, Green JB, Berg T, Norum KR. Vitamin A metabolism: new perspectives on absorption, transport, and storage. Physiol Rev. 1991;71:951–990. - PubMed
-
- Chen N, Napoli JL. All-trans-retinoic acid stimulates translation and induces spine formation in hippocampal neurons through a membrane-associated RARalpha. Faseb J. 2008;22:236–245. - PubMed
-
- Chiang MY, Misner D, Kempermann G, Schikorski T, Giguere V, Sucov HM, Gage FH, Stevens CF, Evans RM. An essential role for retinoid receptors RARbeta and RXRgamma in long-term potentiation and depression. Neuron. 1998;21:1353–1361. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
