

Screening Outside the Catalytic Site: Inhibition of Macromolecular Inter-actions Through Structure-Based Virtual Ligand Screening Experiments
- PMID: 18949072
- PMCID: PMC2570552
- DOI: 10.2174/1874091X00802010029
Screening Outside the Catalytic Site: Inhibition of Macromolecular Inter-actions Through Structure-Based Virtual Ligand Screening Experiments
Abstract
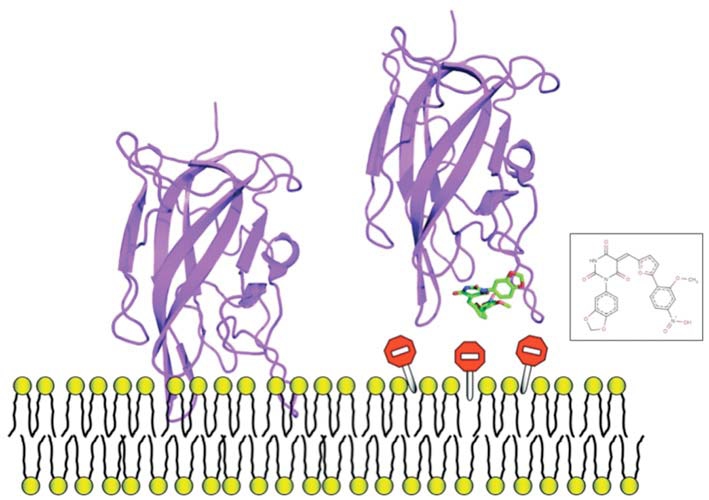
During these last 15 years, drug discovery strategies have essentially focused on identifying small molecules able to inhibit catalytic sites. However, other mechanisms could be targeted. Protein-protein interactions play crucial roles in a number of biological processes, and, as such, their disruption or stabilization is becoming an area of intense activity. Along the same line, inhibition of protein-membrane could be of major importance in several disease indications. Despite the many challenges associated with the development of such classes of interaction modulators, there has been considerable success in the recent years. Importantly, through the existence of protein hot-spots and the presence of druggable pockets at the macromolecular interfaces or in their vicinities, it has been possible to find small molecule effectors using a variety of screening techniques, including combined virtual ligand-in vitro screening strategy. Indeed such in silico-in vitro protocols emerge as the method of choice to facilitate our quest of novel drug-like compounds or of mechanistic probes aiming at facilitating the understanding of molecular reactions involved in the Health and Disease process. In this review, we comment recent successes of combined in silico-in vitro screening methods applied to modulating macromolecular interactions with a special emphasis on protein-membrane interactions.
Keywords: Virtual screening; drug discovery; protein-membrane interaction; protein-protein interaction; structure-based drug design.
Figures
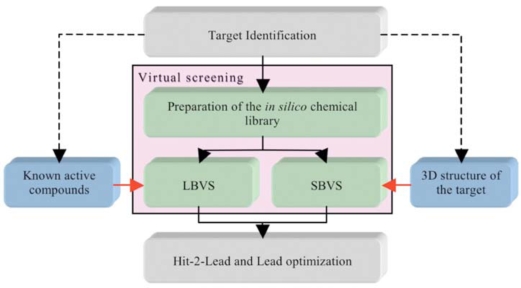
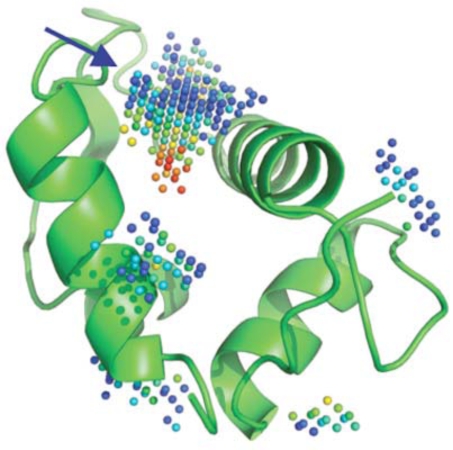
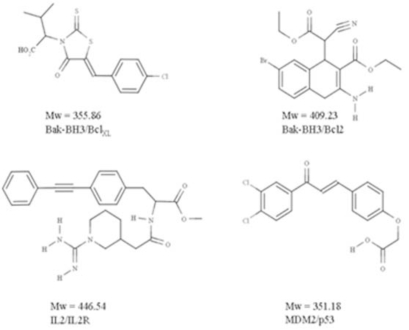
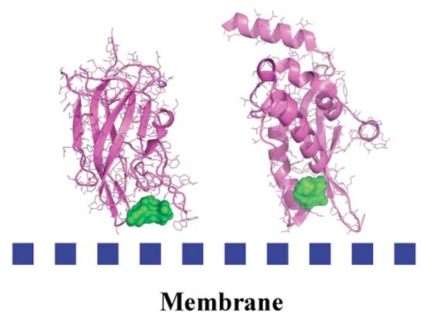
Similar articles
-
Macromolecular crowding: chemistry and physics meet biology (Ascona, Switzerland, 10-14 June 2012).Phys Biol. 2013 Aug;10(4):040301. doi: 10.1088/1478-3975/10/4/040301. Epub 2013 Aug 2. Phys Biol. 2013. PMID: 23912807
-
Design of protein membrane interaction inhibitors by virtual ligand screening, proof of concept with the C2 domain of factor V.Proc Natl Acad Sci U S A. 2007 Jul 31;104(31):12697-702. doi: 10.1073/pnas.0701051104. Epub 2007 Jul 23. Proc Natl Acad Sci U S A. 2007. PMID: 17646652 Free PMC article.
-
Protein-protein interaction inhibitors: small molecules from screening techniques.Curr Top Med Chem. 2007;7(10):922-7. doi: 10.2174/156802607780906735. Curr Top Med Chem. 2007. PMID: 17508923 Review.
-
Free resources to assist structure-based virtual ligand screening experiments.Curr Protein Pept Sci. 2007 Aug;8(4):381-411. doi: 10.2174/138920307781369391. Curr Protein Pept Sci. 2007. PMID: 17696871 Review.
-
In silico-in vitro screening of protein-protein interactions: towards the next generation of therapeutics.Curr Pharm Biotechnol. 2008 Apr;9(2):103-22. doi: 10.2174/138920108783955218. Curr Pharm Biotechnol. 2008. PMID: 18393867 Review.
Cited by
-
Druggable orthosteric and allosteric hot spots to target protein-protein interactions.Curr Pharm Des. 2014;20(8):1293-301. doi: 10.2174/13816128113199990073. Curr Pharm Des. 2014. PMID: 23713780 Free PMC article. Review.
-
Proline-Rich Protein Tyrosine Kinase 2 in Inflammation and Cancer.Cancers (Basel). 2018 May 8;10(5):139. doi: 10.3390/cancers10050139. Cancers (Basel). 2018. PMID: 29738483 Free PMC article. Review.
-
Integrative computational protocol for the discovery of inhibitors of the Helicobacter pylori nickel response regulator (NikR).J Mol Model. 2011 Dec;17(12):3075-84. doi: 10.1007/s00894-011-0962-2. Epub 2011 Mar 1. J Mol Model. 2011. PMID: 21360181
-
Computer applications for prediction of protein-protein interactions and rational drug design.Adv Appl Bioinform Chem. 2009;2:101-23. Epub 2009 Nov 10. Adv Appl Bioinform Chem. 2009. PMID: 21918619 Free PMC article.
-
A single-domain llama antibody potently inhibits the enzymatic activity of botulinum neurotoxin by binding to the non-catalytic alpha-exosite binding region.J Mol Biol. 2010 Apr 9;397(4):1106-18. doi: 10.1016/j.jmb.2010.01.070. Epub 2010 Feb 6. J Mol Biol. 2010. PMID: 20138889 Free PMC article.
References
-
- Whitty A, Kumaravel G. Nat. Chem. Biol. 2006;2:112–118. - PubMed
-
- Block P, Weskamp N, Wolf A, Klebe G. Proteins. 2007;68:170–186. - PubMed
-
- Hajduk PJ, Huth JR, Tse C. Drug Discov. Today. 2005;10:1675–1682. - PubMed
-
- Gaither LA. Expert Rev. Proteomics. 2007;4:411–419. - PubMed
-
- Harris CJ, Stevens A.P. Drug Discov. Today. 2006;11:880–888. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources