

Tumor necrosis factor-alpha potentiates intraneuronal Ca2+ signaling via regulation of the inositol 1,4,5-trisphosphate receptor
- PMID: 18838384
- PMCID: PMC2586262
- DOI: 10.1074/jbc.M802209200
Tumor necrosis factor-alpha potentiates intraneuronal Ca2+ signaling via regulation of the inositol 1,4,5-trisphosphate receptor
Abstract
Inflammatory events have long been implicated in initiating and/or propagating the pathophysiology associated with a number of neurological diseases. In addition, defects in Ca2+-handling processes, which shape membrane potential, influence gene transcription, and affect neuronal spiking patterns, have also been implicated in disease progression and cognitive decline. The mechanisms underlying the purported interplay that exists between neuroinflammation and Ca2+ homeostasis have yet to be defined. Herein, we describe a novel neuron-intrinsic pathway in which the expression of the type-1 inositol 1,4,5-trisphosphate receptor is regulated by the potent pro-inflammatory cytokine tumor necrosis factor-alpha. Exposure of primary murine neurons to tumor necrosis factor-alpha resulted in significant enhancement of Ca2+ signals downstream of muscarinic and purinergic stimulation. An increase in type-1 inositol 1,4,5-trisphosphate receptor mRNA and protein steady-state levels following cytokine exposure positively correlated with this alteration in Ca2+ homeostasis. Modulation of Ca2+ responses arising from this receptor subtype and its downstream effectors may exact significant consequences on neuronal function and could underlie the compromise in neuronal activity observed in the setting of chronic neuroinflammation, such as that associated with Parkinson disease and Alzheimer disease.
Figures
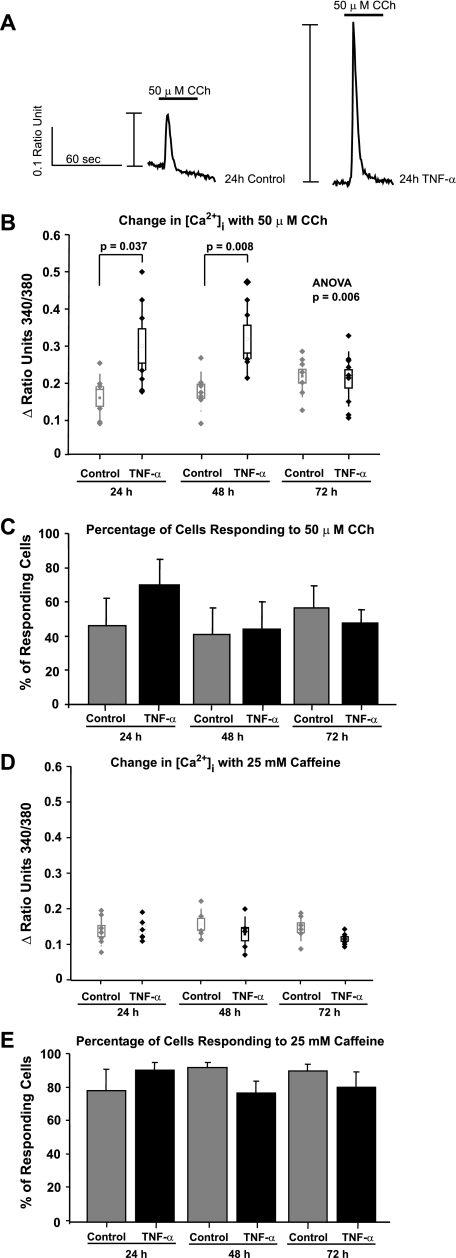
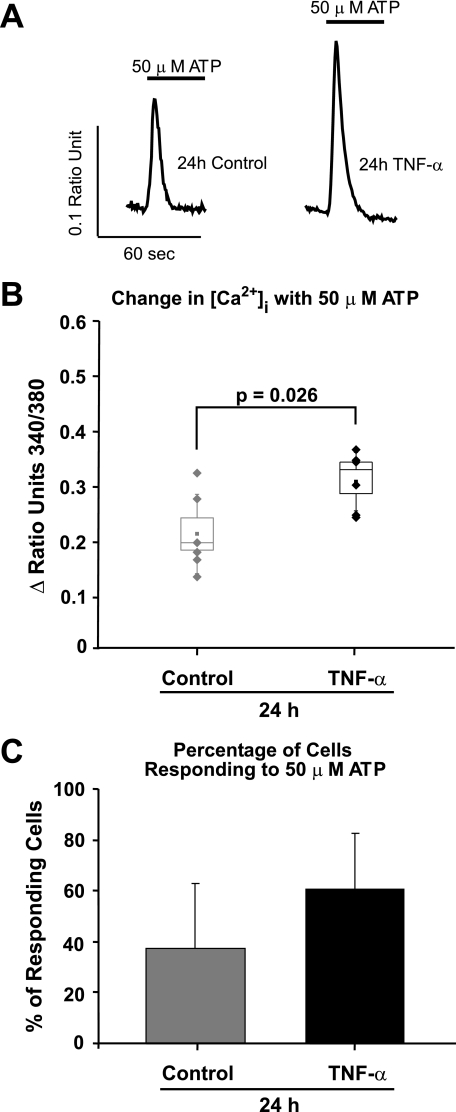
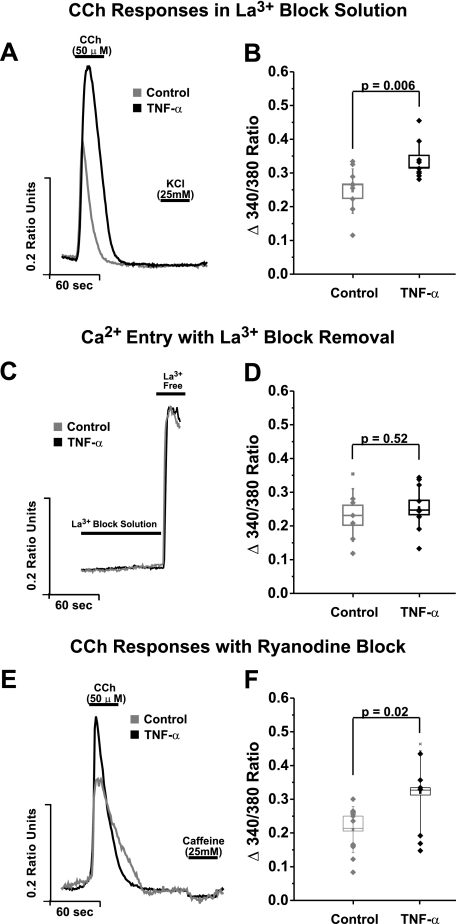
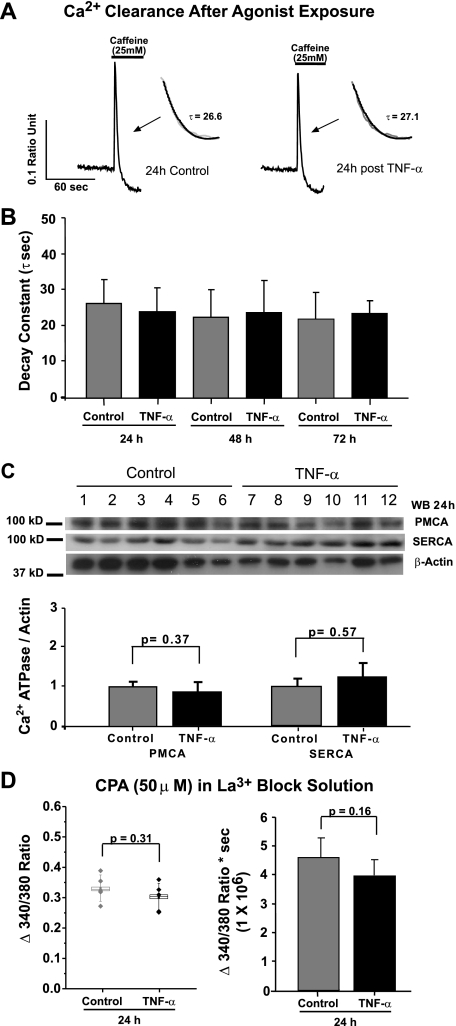
Similar articles
-
Tumor necrosis factor-alpha-mediated regulation of the inositol 1,4,5-trisphosphate receptor promoter.J Biol Chem. 2009 Oct 2;284(40):27557-66. doi: 10.1074/jbc.M109.034504. Epub 2009 Aug 7. J Biol Chem. 2009. PMID: 19666470 Free PMC article.
-
Impaired TNF-alpha control of IP3R-mediated Ca2+ release in Alzheimer's disease mouse neurons.Cell Signal. 2010 Mar;22(3):519-26. doi: 10.1016/j.cellsig.2009.11.006. Cell Signal. 2010. PMID: 19922794 Free PMC article.
-
Modulation of Gq-protein-coupled inositol trisphosphate and Ca2+ signaling by the membrane potential.J Neurosci. 2006 Sep 27;26(39):9983-95. doi: 10.1523/JNEUROSCI.2773-06.2006. J Neurosci. 2006. PMID: 17005862 Free PMC article.
-
Modulation of cytosolic calcium signaling by protein kinase A-mediated phosphorylation of inositol 1,4,5-trisphosphate receptors.Biol Res. 2004;37(4):593-602. doi: 10.4067/s0716-97602004000400013. Biol Res. 2004. PMID: 15709686 Review.
-
Inositol trisphosphate receptor Ca2+ release channels in neurological diseases.Pflugers Arch. 2010 Jul;460(2):481-94. doi: 10.1007/s00424-010-0826-0. Epub 2010 Apr 10. Pflugers Arch. 2010. PMID: 20383523 Free PMC article. Review.
Cited by
-
Calcium dysregulation and neuroinflammation: discrete and integrated mechanisms for age-related synaptic dysfunction.Ageing Res Rev. 2013 Sep;12(4):982-95. doi: 10.1016/j.arr.2013.05.008. Epub 2013 Jun 8. Ageing Res Rev. 2013. PMID: 23751484 Free PMC article. Review.
-
Tumor necrosis factor-alpha-induced reduction of glomerular filtration rate in rats with fulminant hepatic failure.Lab Invest. 2014 Jul;94(7):740-51. doi: 10.1038/labinvest.2014.71. Epub 2014 Jun 2. Lab Invest. 2014. PMID: 24887412
-
Scutellarin as a Potential Therapeutic Agent for Microglia-Mediated Neuroinflammation in Cerebral Ischemia.Neuromolecular Med. 2016 Sep;18(3):264-73. doi: 10.1007/s12017-016-8394-x. Epub 2016 Apr 21. Neuromolecular Med. 2016. PMID: 27103430 Review.
-
Tumor necrosis factor-alpha-mediated regulation of the inositol 1,4,5-trisphosphate receptor promoter.J Biol Chem. 2009 Oct 2;284(40):27557-66. doi: 10.1074/jbc.M109.034504. Epub 2009 Aug 7. J Biol Chem. 2009. PMID: 19666470 Free PMC article.
-
The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia.J Neuroinflammation. 2010 Nov 1;7:74. doi: 10.1186/1742-2094-7-74. J Neuroinflammation. 2010. PMID: 21040547 Free PMC article. Review.
References
-
- McGeer, E. G., Klegeris, A., and McGeer, P. L. (2005) Neurobiol. Aging. 26 Suppl. 1, 94–97 - PubMed
-
- Ortaldo, J. R., Mason, L. H., Mathieson, B. J., Liang, S. M., Flick, D. A., and Herberman, R. B. (1986) Nature 321 700–702 - PubMed
-
- Laster, S. M., Wood, J. G., and Gooding, L. R. (1988) J. Immunol. 141 2629–2634 - PubMed
-
- Hanisch, U. K. (2002) Glia 40 140–155 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
