

Prader-Willi syndrome
- PMID: 18781185
- PMCID: PMC2985966
- DOI: 10.1038/ejhg.2008.165
Prader-Willi syndrome
Abstract
Prader-Willi syndrome (PWS) is a highly variable genetic disorder affecting multiple body systems whose most consistent major manifestations include hypotonia with poor suck and poor weight gain in infancy; mild mental retardation, hypogonadism, growth hormone insufficiency causing short stature for the family, early childhood-onset hyperphagia and obesity, characteristic appearance, and behavioral and sometimes psychiatric disturbance. Many more minor characteristics can be helpful in diagnosis and important in management. PWS is an example of a genetic condition involving genomic imprinting. It can occur by three main mechanisms, which lead to absence of expression of paternally inherited genes in the 15q11.2-q13 region: paternal microdeletion, maternal uniparental disomy, and imprinting defect.
Figures


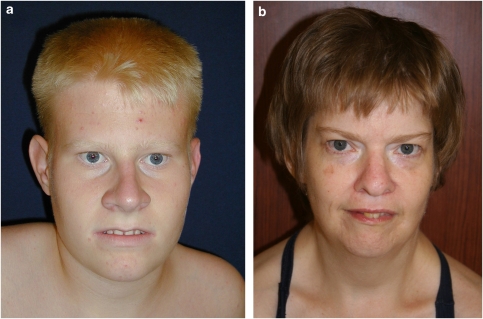

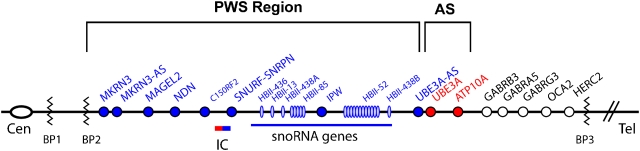

Comment in
-
Prader-Willi and Angelman syndromes: genetic counseling.Eur J Hum Genet. 2010 Feb;18(2):154-5; author reply 155-6. doi: 10.1038/ejhg.2009.170. Epub 2009 Oct 7. Eur J Hum Genet. 2010. PMID: 19809481 Free PMC article. No abstract available.
Similar articles
-
The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria.Pediatrics. 2001 Nov;108(5):E92. doi: 10.1542/peds.108.5.e92. Pediatrics. 2001. PMID: 11694676
-
Prader-Willi Syndrome - Clinical Genetics, Diagnosis and Treatment Approaches: An Update.Curr Pediatr Rev. 2019;15(4):207-244. doi: 10.2174/1573396315666190716120925. Curr Pediatr Rev. 2019. PMID: 31333129 Free PMC article. Review.
-
Prader-Willi Syndrome: Clinical Genetics and Diagnostic Aspects with Treatment Approaches.Curr Pediatr Rev. 2016;12(2):136-66. doi: 10.2174/1573396312666151123115250. Curr Pediatr Rev. 2016. PMID: 26592417 Free PMC article. Review.
-
Relationship of thyroid function with genetic subtypes and treatment with growth hormone in Prader-Willi syndrome.Am J Med Genet A. 2024 Oct;194(10):e63724. doi: 10.1002/ajmg.a.63724. Epub 2024 Jun 4. Am J Med Genet A. 2024. PMID: 38837660
-
Prader-Willi syndrome.Genet Med. 2012 Jan;14(1):10-26. doi: 10.1038/gim.0b013e31822bead0. Epub 2011 Sep 26. Genet Med. 2012. PMID: 22237428 Review.
Cited by
-
Copy number variants in obesity-related syndromes: review and perspectives on novel molecular approaches.J Obes. 2012;2012:845480. doi: 10.1155/2012/845480. Epub 2012 Dec 17. J Obes. 2012. PMID: 23316347 Free PMC article. Review.
-
Firing activity of locus coeruleus noradrenergic neurons decreases in necdin-deficient mice, an animal model of Prader-Willi syndrome.J Neurodev Disord. 2020 Jul 29;12(1):21. doi: 10.1186/s11689-020-09323-4. J Neurodev Disord. 2020. PMID: 32727346 Free PMC article.
-
Complex Economic Behavior Patterns Are Constructed from Finite, Genetically Controlled Modules of Behavior.Cell Rep. 2019 Aug 13;28(7):1814-1829.e6. doi: 10.1016/j.celrep.2019.07.038. Cell Rep. 2019. PMID: 31412249 Free PMC article.
-
Free Insulin-like Growth Factor (IGF)-I in Children with PWS.J Clin Med. 2022 Feb 26;11(5):1280. doi: 10.3390/jcm11051280. J Clin Med. 2022. PMID: 35268371 Free PMC article.
-
Repetitive behavior profiles: Consistency across autism spectrum disorder cohorts and divergence from Prader-Willi syndrome.J Neurodev Disord. 2011 Dec;3(4):316-24. doi: 10.1007/s11689-011-9094-3. Epub 2011 Sep 1. J Neurodev Disord. 2011. PMID: 21881965 Free PMC article.
References
-
- Miller SP, Riley P, Shevell MI. The neonatal presentation of Prader-Willi syndrome revisited. J Pediatr. 1999;134:226–228. - PubMed
-
- Gunay-Aygun M, Schwartz S, O'Riordan MA, Cassidy SB. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics. 2001;108:E92. - PubMed
-
- Crinó A, Schiaffini R, Ciampalini P, et al. Hypogonadism and pubertal development in Prader-Willi syndrome. Eur J Pediatr. 2003;162:327–333. - PubMed
-
- Dykens EM, Hodapp RM, Walsh K, Nash L. Profiles, correlates and trajectories of intelligence in individuals with Prader-Willi syndrome. J Am Acad Child Adolesc Psychiatry. 1992;31:1125–1130. - PubMed
-
- Curfs LM, Fryns JP. Prader-Willi syndrome: a review with special attention to the cognitive and behavioral profile. Birth Defects Orig Artic Ser. 1992;28:99–104. - PubMed
Further Reading
-
- Cassidy SB, Schwartz S.Prader-Willi Syndrome GeneReviewsCopyright, Seattle: University of Washington; updated March 2008. http://www.genetests.org .
-
- Goldstone AP. Prader-Willi syndrome: advances in genetics, pathophysiology and treatment. Trends Endocrinol Metab. 2004;15:12–20. - PubMed
-
- McCandless SE, Cassidy SB.15q11-13 and the Prader-Willi syndromein: Epstein CJ, Erickson RP, Wynshaw-Boris A (eds): Molecular Basis of Inborn Errors of Development Oxford University Press; 2008. 3rd edn, ch. 105.
-
- Prader-Willi Syndrome Association (USA) website: www.pwsausa.org .
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
