

Monte carlo simulations of protein assembly, disassembly, and linear motion on DNA
- PMID: 18658217
- PMCID: PMC2576399
- DOI: 10.1529/biophysj.108.135061
Monte carlo simulations of protein assembly, disassembly, and linear motion on DNA
Abstract
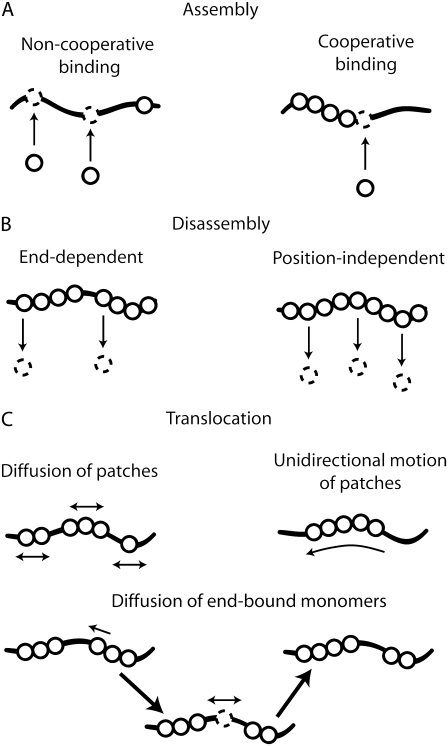
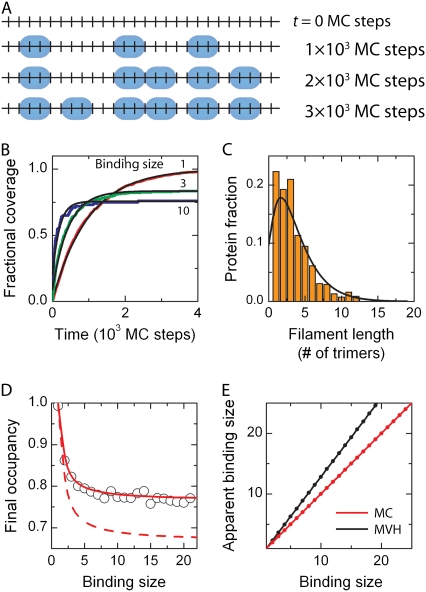
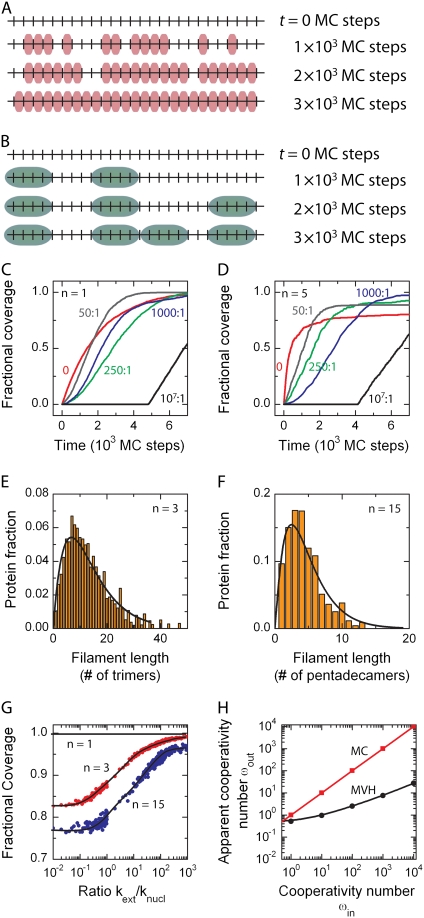
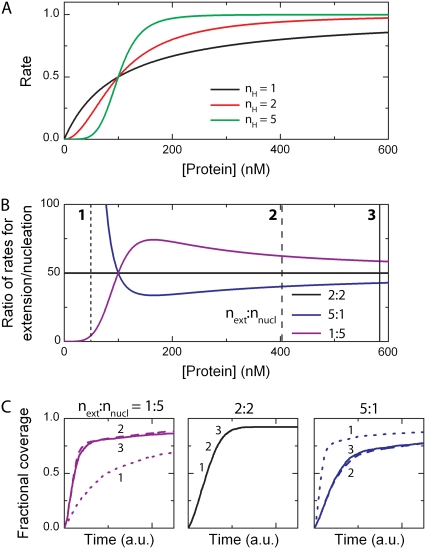
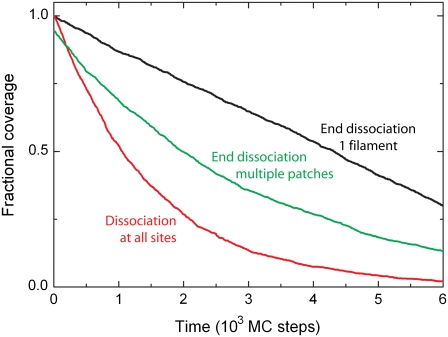
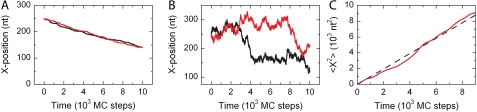
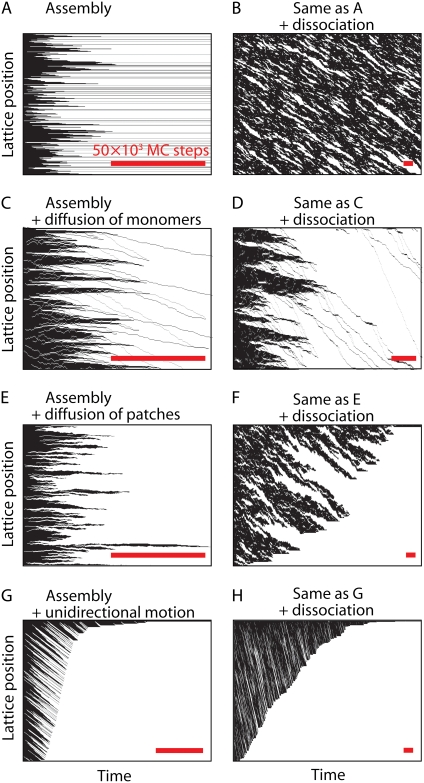
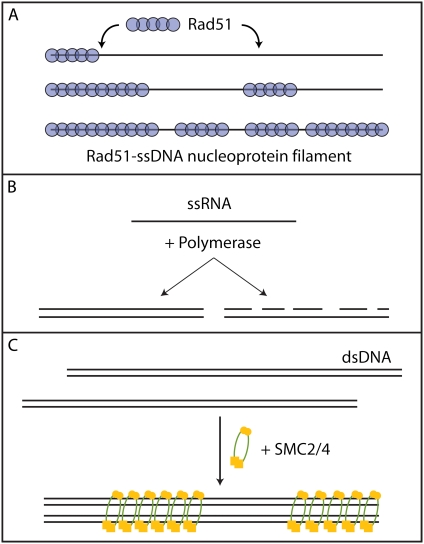
We use Monte Carlo simulations to analyze the simultaneous interactions of multiple proteins to a long DNA molecule. We study the time dependence of protein organization on DNA for different regimes that comprise (non)cooperative sequence-independent protein assembly, dissociation, and linear motion. A range of different behaviors is observed for the dynamics, final coverage, and cluster size distributions. We observe that the DNA substrate is almost never completely covered by protein when taking into account only (non)cooperative binding, because gaps remain on the substrate that are smaller than the binding site size of the protein. Due to these gaps, the apparent binding size of a protein during noncooperative binding can be overestimated by up to 30%. During dissociation of cooperatively bound proteins, the dissociation curve can be exponentially shaped even when allowing only end-dependent dissociation. We discuss the potential of our method for the analysis of a number of single-molecule experiments, for example, the binding of the DNA-repair proteins RecA and Rad51 to DNA.
Figures
Similar articles
-
Geometry of mediating protein affects the probability of loop formation in DNA.Biophys J. 2008 Apr 15;94(8):3150-8. doi: 10.1529/biophysj.107.122986. Epub 2008 Jan 11. Biophys J. 2008. PMID: 18192346 Free PMC article.
-
Protein sliding and hopping kinetics on DNA.Phys Rev E Stat Nonlin Soft Matter Phys. 2011 Feb;83(2 Pt 1):021907. doi: 10.1103/PhysRevE.83.021907. Epub 2011 Feb 16. Phys Rev E Stat Nonlin Soft Matter Phys. 2011. PMID: 21405863 Free PMC article.
-
Discrete persistent-chain model for protein binding on DNA.Phys Rev E Stat Nonlin Soft Matter Phys. 2011 Apr;83(4 Pt 1):041912. doi: 10.1103/PhysRevE.83.041912. Epub 2011 Apr 18. Phys Rev E Stat Nonlin Soft Matter Phys. 2011. PMID: 21599206
-
An introduction to the mechanics of DNA.Philos Trans A Math Phys Eng Sci. 2004 Jul 15;362(1820):1265-79. doi: 10.1098/rsta.2004.1392. Philos Trans A Math Phys Eng Sci. 2004. PMID: 15306450 Review.
-
The structural basis of DNA flexibility.Philos Trans A Math Phys Eng Sci. 2004 Jul 15;362(1820):1423-38. doi: 10.1098/rsta.2004.1390. Philos Trans A Math Phys Eng Sci. 2004. PMID: 15306459 Review.
Cited by
-
Visualization and quantification of nascent RAD51 filament formation at single-monomer resolution.Proc Natl Acad Sci U S A. 2014 Oct 21;111(42):15090-5. doi: 10.1073/pnas.1307824111. Epub 2014 Oct 6. Proc Natl Acad Sci U S A. 2014. PMID: 25288749 Free PMC article.
-
Effect of the BRCA2 CTRD domain on RAD51 filaments analyzed by an ensemble of single molecule techniques.Nucleic Acids Res. 2011 Aug;39(15):6558-67. doi: 10.1093/nar/gkr295. Epub 2011 May 16. Nucleic Acids Res. 2011. PMID: 21576230 Free PMC article.
-
Single-nucleoid architecture reveals heterogeneous packaging of mitochondrial DNA.Nat Struct Mol Biol. 2024 Mar;31(3):568-577. doi: 10.1038/s41594-024-01225-6. Epub 2024 Feb 12. Nat Struct Mol Biol. 2024. PMID: 38347148
-
Torsional regulation of hRPA-induced unwinding of double-stranded DNA.Nucleic Acids Res. 2010 Jul;38(12):4133-42. doi: 10.1093/nar/gkq067. Epub 2010 Mar 2. Nucleic Acids Res. 2010. PMID: 20197317 Free PMC article.
-
Dynamics of RecA filaments on single-stranded DNA.Nucleic Acids Res. 2009 Jul;37(12):4089-99. doi: 10.1093/nar/gkp326. Epub 2009 May 8. Nucleic Acids Res. 2009. PMID: 19429893 Free PMC article.
References
-
- Seidel, R., J. van Noort, C. van der Scheer, J. G. P. Bloom, N. H. Dekker, C. F. Dutta, A. Blundell, T. Robinson, K. Firman, and C. Dekker. 2004. Real-time observation of DNA translocation by the type I restriction modification enzyme EcoR124I. Nat. Struct. Mol. Biol. 11:838–843. - PubMed
-
- McGhee, J. D., and P. H. von Hippel. 1974. Theoretical aspects of DNA-protein interactions—cooperative and non-cooperative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 86:469–489. - PubMed
-
- Lonberg, N., S. C. Kowalczykowski, L. S. Paul, and P. H. von Hippel. 1981. Interactions of bacteriophage T4-coded gene 32 protein with nucleic acids: III. Binding-properties of 2 specific proteolytic digestion products of the protein (G32P*I and G32P*III). J. Mol. Biol. 145:123–138. - PubMed
-
- Ando, R. A., and S. W. Morrical. 1998. Single-stranded DNA binding properties of the UvsX recombinase of bacteriophage T4: binding parameters and effects of nucleotides. J. Mol. Biol. 283:785–796. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
