

Oxidative stress in Fanconi anemia hematopoiesis and disease progression
- PMID: 18627348
- PMCID: PMC2695607
- DOI: 10.1089/ars.2008.2129
Oxidative stress in Fanconi anemia hematopoiesis and disease progression
Abstract
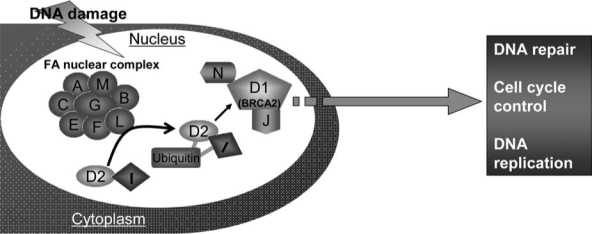
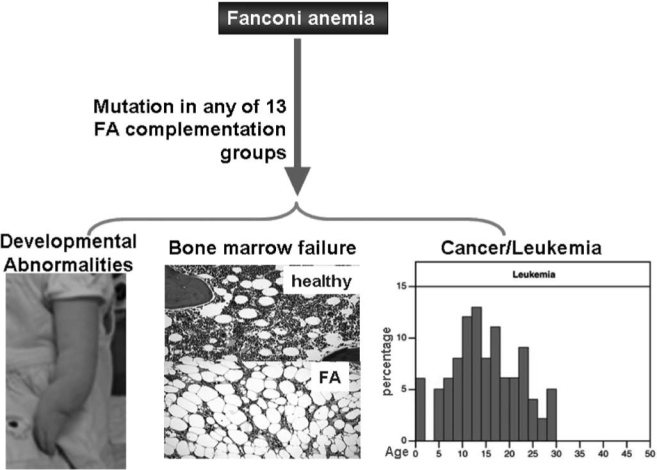
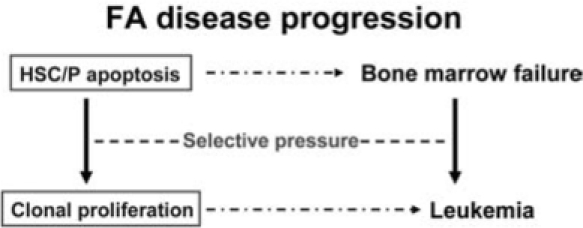
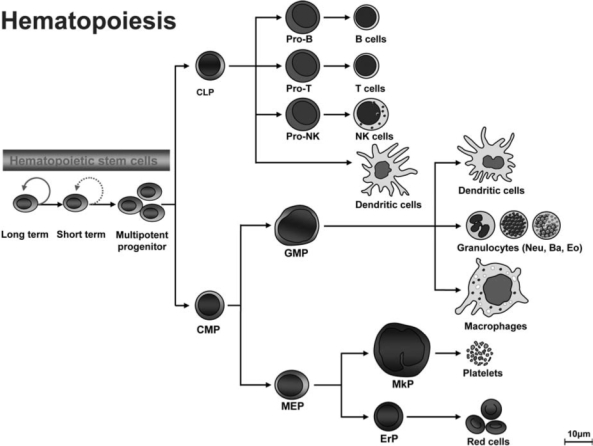
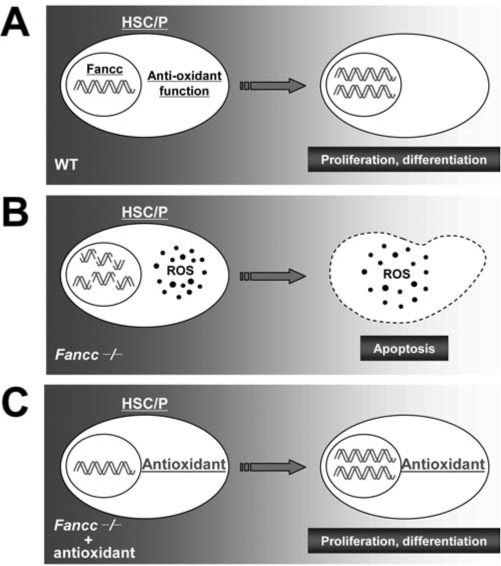
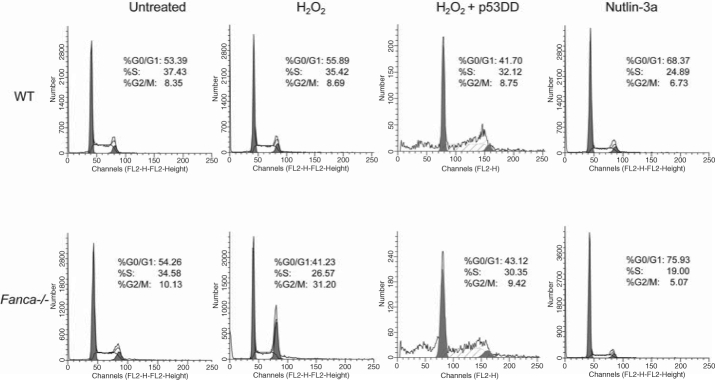
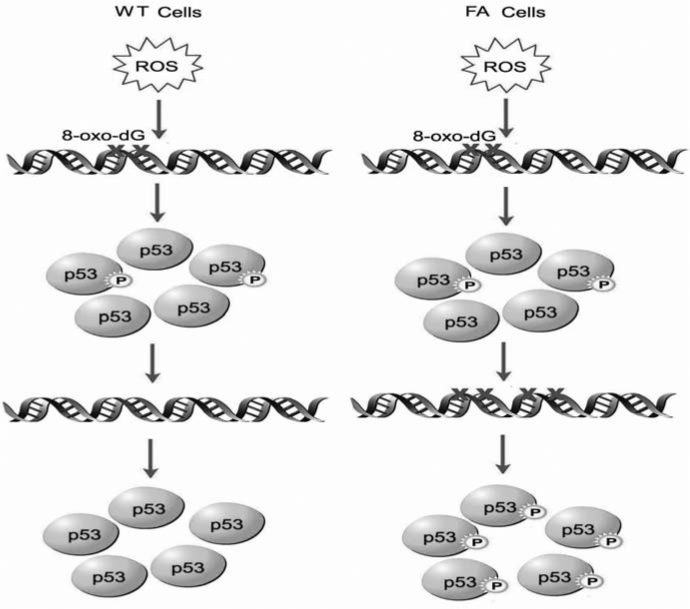
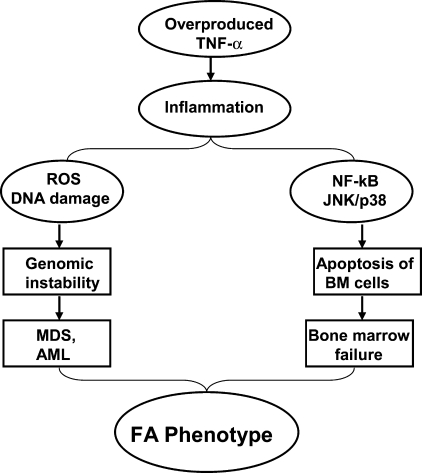
Patients with the genomic instability syndrome Fanconi anemia (FA) commonly develop progressive bone marrow failure and have a high risk of cancer. The prominent role of the FA protein family involves DNA damage response and/or repair. Oxidative stress, defined as an imbalance between the production of reactive oxygen species and antioxidant defense, is considered to be an important pathogenic factor in leukemia-prone bone marrow diseases such as FA. Cellular responses inducing resistance to oxidative stress are important for cellular survival, organism lifespan, and cancer prevention, but until recently, mammalian factors regulating resistance to oxidative stress have not been well characterized. Significant evidence supports excessive apoptosis of hematopoietic stem/progenitor cells, induced by stresses, most significantly oxidative stress, as a critical factor in the pathogenesis of bone marrow failure and leukemia progression in FA. In this brief review, we discuss the functional link between FA proteins and oxidative DNA damage response/repair, with emphasis on the implication of oxidative stress in the pathophysiology and abnormal hematopoiesis in FA.
Figures
Similar articles
-
TNF-α signaling in Fanconi anemia.Blood Cells Mol Dis. 2014 Jan;52(1):2-11. doi: 10.1016/j.bcmd.2013.06.005. Epub 2013 Jul 24. Blood Cells Mol Dis. 2014. PMID: 23890415 Free PMC article. Review.
-
Current knowledge on the pathophysiology of Fanconi anemia: from genes to phenotypes.Int J Hematol. 2001 Jul;74(1):33-41. doi: 10.1007/BF02982547. Int J Hematol. 2001. PMID: 11530803 Review.
-
Persistent response of Fanconi anemia haematopoietic stem and progenitor cells to oxidative stress.Cell Cycle. 2017 Jun 18;16(12):1201-1209. doi: 10.1080/15384101.2017.1320627. Epub 2017 May 5. Cell Cycle. 2017. PMID: 28475398 Free PMC article.
-
The FA pathway counteracts oxidative stress through selective protection of antioxidant defense gene promoters.Blood. 2012 May 3;119(18):4142-51. doi: 10.1182/blood-2011-09-381970. Epub 2012 Mar 9. Blood. 2012. PMID: 22408259 Free PMC article.
-
[Fanconi anemia: cellular and molecular features].Pathol Biol (Paris). 2007 Feb;55(1):19-28. doi: 10.1016/j.patbio.2006.04.008. Epub 2006 Aug 10. Pathol Biol (Paris). 2007. PMID: 16904272 Review. French.
Cited by
-
Salidroside stimulates DNA repair enzyme Parp-1 activity in mouse HSC maintenance.Blood. 2012 May 3;119(18):4162-73. doi: 10.1182/blood-2011-10-387332. Epub 2012 Mar 16. Blood. 2012. PMID: 22427203 Free PMC article.
-
Proteomic profiling and bioinformatics analysis identify key regulators during the process from fanconi anemia to acute myeloid leukemia.Am J Transl Res. 2020 Apr 15;12(4):1415-1427. eCollection 2020. Am J Transl Res. 2020. PMID: 32355551 Free PMC article.
-
Effects of Deacetylase Inhibition on the Activation of the Antioxidant Response and Aerobic Metabolism in Cellular Models of Fanconi Anemia.Antioxidants (Basel). 2023 May 15;12(5):1100. doi: 10.3390/antiox12051100. Antioxidants (Basel). 2023. PMID: 37237966 Free PMC article.
-
Mesenchymal stem/stromal cells from a transplanted, asymptomatic patient with Fanconi anemia exhibit an aging-like phenotype and dysregulated expression of genes implicated in hematopoiesis and myelodysplasia.Cytotherapy. 2023 Apr;25(4):362-368. doi: 10.1016/j.jcyt.2022.11.003. Epub 2022 Dec 5. Cytotherapy. 2023. PMID: 36481320 Free PMC article.
-
Metformin ameliorates ionizing irradiation-induced long-term hematopoietic stem cell injury in mice.Free Radic Biol Med. 2015 Oct;87:15-25. doi: 10.1016/j.freeradbiomed.2015.05.045. Epub 2015 Jun 16. Free Radic Biol Med. 2015. PMID: 26086617 Free PMC article.
References
-
- Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nature Rev Immunol. 2003;3:745–756. - PubMed
-
- Bagby GC., Jr Genetic basis of Fanconi anemia. Curr Opin Hematol. 2003;10:68–76. - PubMed
-
- Balkwill F. Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. - PubMed
-
- Bogliolo M. Cabré O. Callén E. Castillo V. Creus A. Marcos R. Surrallés J. The Fanconi anaemia genome stability and tumour suppressor network. Mutagenesis. 2002;17:529–538. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
