

Review
doi: 10.1038/nrm2450.
Epub 2008 Jul 2.
ATR: an essential regulator of genome integrity
Affiliations
- PMID: 18594563
- PMCID: PMC2663384
- DOI: 10.1038/nrm2450
Item in Clipboard
Review
ATR: an essential regulator of genome integrity
Nat Rev Mol Cell Biol.
2008 Aug.
Abstract
Genome maintenance is a constant concern for cells, and a coordinated response to DNA damage is required to maintain cellular viability and prevent disease. The ataxia-telangiectasia mutated (ATM) and ATM and RAD3-related (ATR) protein kinases act as master regulators of the DNA-damage response by signalling to control cell-cycle transitions, DNA replication, DNA repair and apoptosis. Recent studies have provided new insights into the mechanisms that control ATR activation, have helped to explain the overlapping but non-redundant activities of ATR and ATM in DNA-damage signalling, and have clarified the crucial functions of ATR in maintaining genome integrity.
Figures
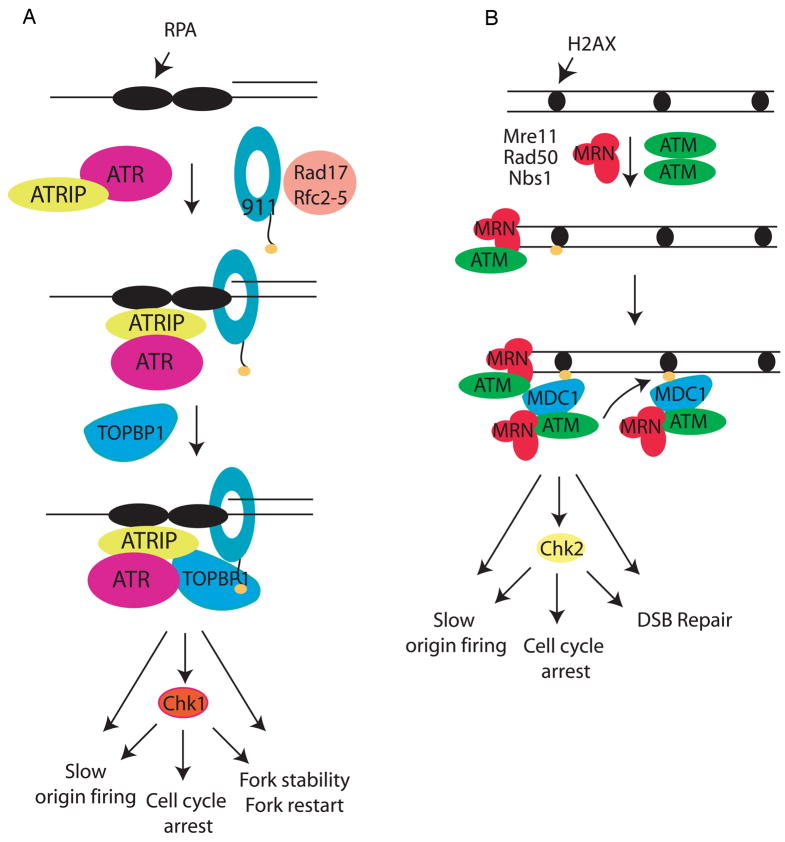
(A) The ATR-ATRIP complex and the 9-1-1 complex are recruited to the ssDNA-5′ primer junction independently. RPA binds ATRIP and directs the Rad17-RFC complex to load the 9-1-1 checkpoint clamp at the 5′ primer junction. Loading of 9-1-1 brings the ATR activator TopBP1 to the damage site through an interaction involving two BRCT domains of TopBP1 and the phosphorylated C-terminal tail of Rad9 (see text). TopBP1 binds and activates ATR in an ATRIP-dependent manner, leading to phosphorylation of the downstream kinase Chk1 and other ATR effectors. In response to DNA damage or replication stress, ATR and its effectors ultimately slow origin firing and induce cell cycle arrest as well as stabilize and restart stalled replication forks. (B) Formation of a double-stranded DNA end leads to recruitment of the MRN complex and dissociation of the dimeric, inactive form of ATM to a monomeric, phosphorylated form. This monomeric form of ATM binds the MRN complex at the DSB, and is further activated by the DNA and MRN complex. Activated ATM then phosphorylates the carboxy terminal tail of the histone variant, H2AX. Phosphorylated H2AX (γ-H2AX) binds to MDC1 through the BRCT domains of MDC1, leading to recruitment of additional ATM/MRN complexes and further H2AX phosphorylation. The activated ATM also phosphorylates downstream targets, among which are Chk2. Phosphorylation of these proteins leads to cell cycle arrest, inhibition of oirgin firing in S phase and double-strand break repair.
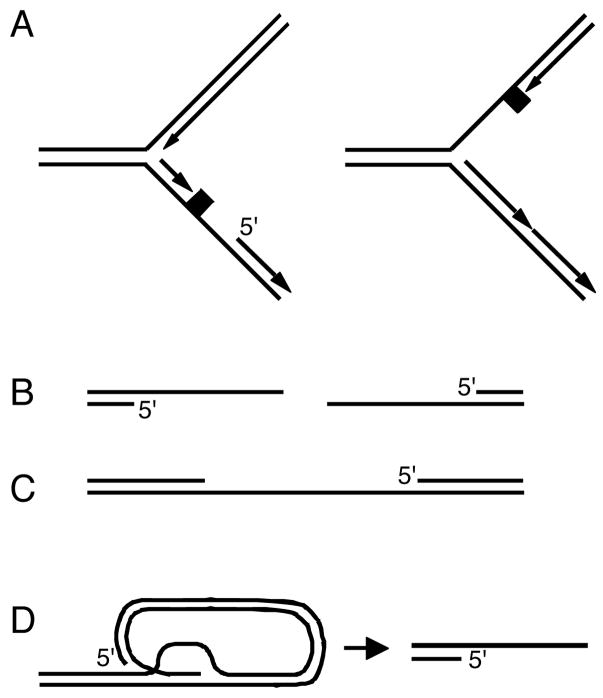
ssDNA gaps with a 5′ primer end are formed during nucleic acid metabolism. Most noteably, helicase/polymerase uncoupling occurs at replication forks upon encountering lesions that stall the polymerase but not the more permissive helicase. A lesion on the lagging strand would immediately leave a gap with a 5′ primer end. Lesions on the leading strand would require repriming to generate the gapped structure. End resection of DSBs and intermediates in nucleotide excision DNA repair also form the ATR activating structure. Finally, telomere erosion such that the normal telomere capping mechanism is removed produces a recessed 5′ end adjacent to telomeric ssDNA.
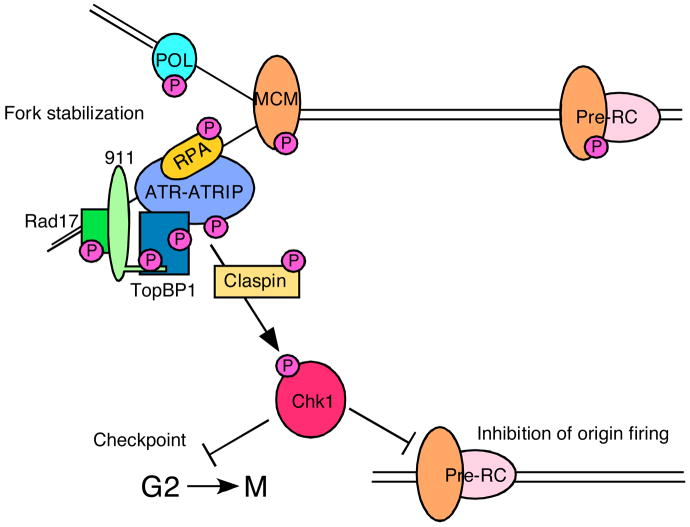
A major ATR substrate is the checkpoint kinase CHK1. CHK1 phosphorylation releases it from chromatin and increases its kinase activity. CHK1 has numerous substrates some of which regulate cell cycle transitions and replication origin firing. Many ATR substrates are at the replication fork. In most cases, the consequences of phosphorylation remain unknown but likely contribute to fork stabilization. As discussed in the text, ATR-dependent MCM2 phosphorylation regulates binding to PLK1 which may promote the completion of DNA replication near the stalled fork.
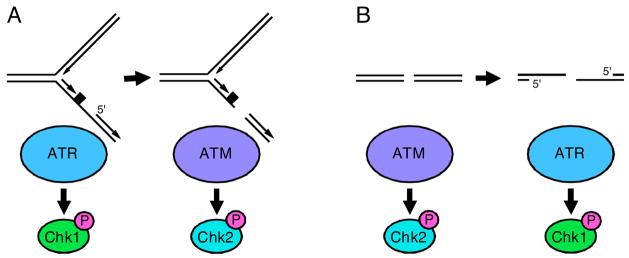
(A) Stalled replication forks activate ATR. Nucleases can cleave stalled forks causing DSBs to form which activate ATM. The rate at which DSBs form at stalled forks is greatly increased in cells with defective ATR signaling. (B) DSBs activate ATM but will also activate ATR as a consequence of DNA end resection. This process is ATM- and cell cycle-dependent such that most ATR activation by double-strand breaks occurs in S and G2 phase cells. CHK1 and CHK2 are primarily ATR and ATM substrates respectively.
Similar articles
-
ATR signalling: more than meeting at the fork.Biochem J. 2011 Jun 15;436(3):527-36. doi: 10.1042/BJ20102162. Biochem J. 2011. PMID: 21615334 Free PMC article. Review.
-
The role of NBS1 in the modulation of PIKK family proteins ATM and ATR in the cellular response to DNA damage.Cancer Lett. 2006 Nov 8;243(1):9-15. doi: 10.1016/j.canlet.2006.01.026. Epub 2006 Mar 10. Cancer Lett. 2006. PMID: 16530324 Free PMC article. Review.
-
Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage.Nature. 2005 Mar 31;434(7033):605-11. doi: 10.1038/nature03442. Epub 2005 Mar 2. Nature. 2005. PMID: 15758953
-
ATM regulates ATR chromatin loading in response to DNA double-strand breaks.J Exp Med. 2006 Feb 20;203(2):297-303. doi: 10.1084/jem.20051923. Epub 2006 Feb 6. J Exp Med. 2006. PMID: 16461339 Free PMC article.
-
Cep164 is a mediator protein required for the maintenance of genomic stability through modulation of MDC1, RPA, and CHK1.Genes Dev. 2008 Mar 1;22(5):587-600. doi: 10.1101/gad.1627708. Epub 2008 Feb 18. Genes Dev. 2008. PMID: 18283122 Free PMC article.
Cited by
-
A TRilogy of ATR's Non-Canonical Roles Throughout the Cell Cycle and Its Relation to Cancer.Cancers (Basel). 2024 Oct 19;16(20):3536. doi: 10.3390/cancers16203536. Cancers (Basel). 2024. PMID: 39456630 Free PMC article. Review.
-
CBP and p300 histone acetyltransferases contribute to homologous recombination by transcriptionally activating the BRCA1 and RAD51 genes.PLoS One. 2012;7(12):e52810. doi: 10.1371/journal.pone.0052810. Epub 2012 Dec 20. PLoS One. 2012. PMID: 23285190 Free PMC article.
-
Autophagy in DNA damage response.Int J Mol Sci. 2015 Jan 23;16(2):2641-62. doi: 10.3390/ijms16022641. Int J Mol Sci. 2015. PMID: 25625517 Free PMC article. Review.
-
Human parvovirus B19: a mechanistic overview of infection and DNA replication.Future Virol. 2015;10(2):155-167. doi: 10.2217/fvl.14.103. Future Virol. 2015. PMID: 26097496 Free PMC article.
-
HUS1 regulates in vivo responses to genotoxic chemotherapies.Oncogene. 2016 Feb 4;35(5):662-9. doi: 10.1038/onc.2015.118. Epub 2015 Apr 27. Oncogene. 2016. PMID: 25915840
References
-
- Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–6. Identified ATRIP and along with reference 129 demonstrated that ATR is essential for cell viability. - PubMed
-
- de Klein A, et al. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol. 2000;10:479–82. - PubMed
-
- Nyberg KA, Michelson RJ, Putnam CW, Weinert TA. Toward maintaining the genome: DNA damage and replication checkpoints. Annu Rev Genet. 2002;36:617–56. - PubMed
-
- Shechter D, Costanzo V, Gautier J. Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair (Amst) 2004;3:901–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
