

A protein functional leap: how a single mutation reverses the function of the transcription regulator TetR
- PMID: 18587152
- PMCID: PMC2490752
- DOI: 10.1093/nar/gkn400
A protein functional leap: how a single mutation reverses the function of the transcription regulator TetR
Abstract
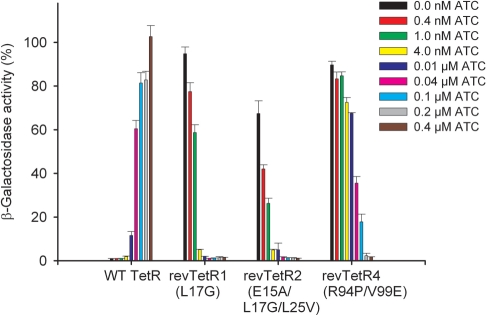
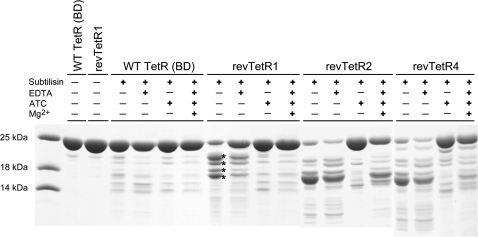
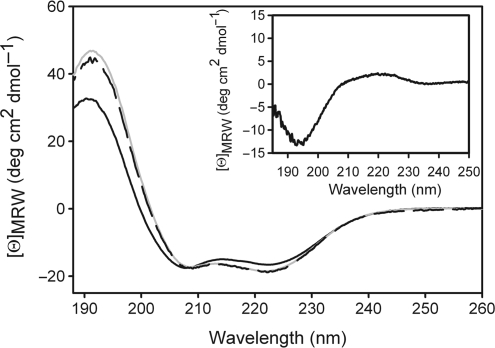
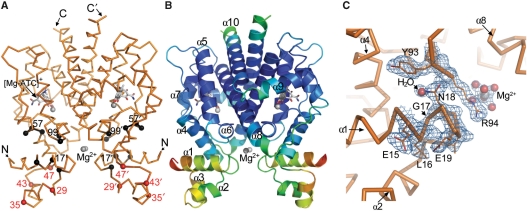
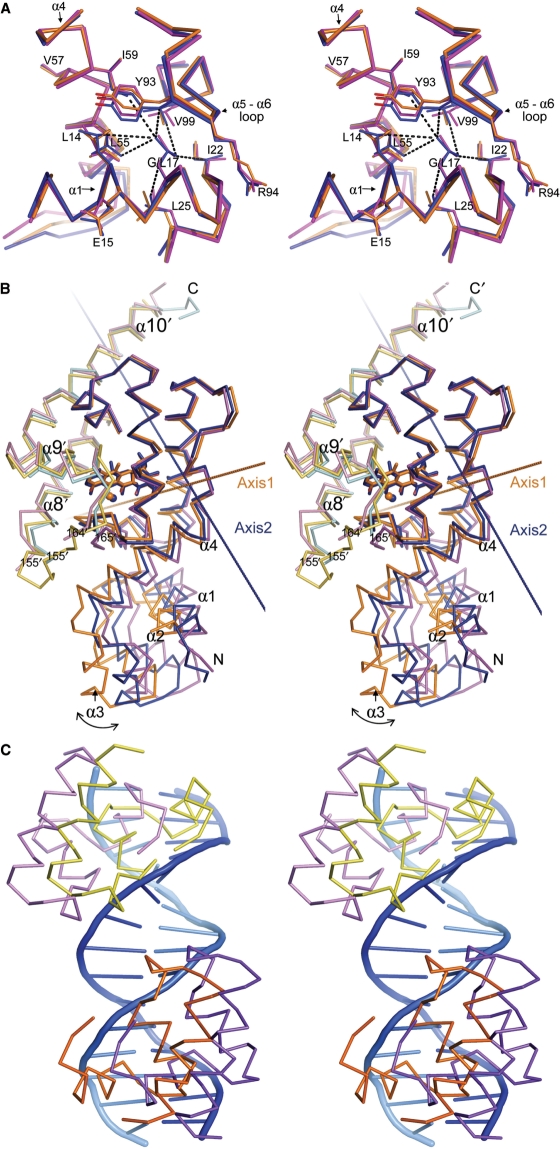
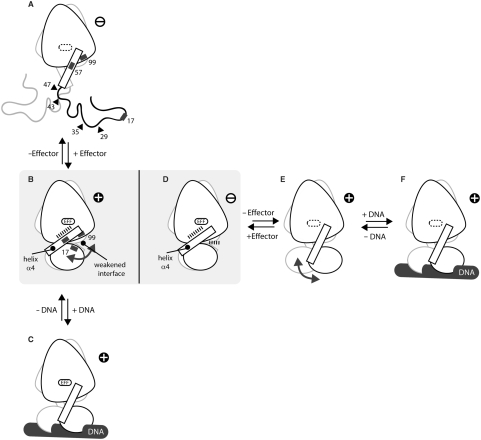
Today's proteome is the result of innumerous gene duplication, mutagenesis, drift and selection processes. Whereas random mutagenesis introduces predominantly only gradual changes in protein function, a case can be made that an abrupt switch in function caused by single amino acid substitutions will not only considerably further evolution but might constitute a prerequisite for the appearance of novel functionalities for which no promiscuous protein intermediates can be envisaged. Recently, tetracycline repressor (TetR) variants were identified in which binding of tetracycline triggers the repressor to associate with and not to dissociate from the operator DNA as in wild-type TetR. We investigated the origin of this activity reversal by limited proteolysis, CD spectroscopy and X-ray crystallography. We show that the TetR mutant Leu17Gly switches its function via a disorder-order mechanism that differs completely from the allosteric mechanism of wild-type TetR. Our study emphasizes how single point mutations can engender unexpected leaps in protein function thus enabling the appearance of new functionalities in proteins without the need for promiscuous intermediates.
Figures
Similar articles
-
Tet repressor induction by tetracycline: a molecular dynamics, continuum electrostatics, and crystallographic study.J Mol Biol. 2008 May 9;378(4):898-912. doi: 10.1016/j.jmb.2008.03.022. Epub 2008 Mar 19. J Mol Biol. 2008. PMID: 18395746
-
Engineered Tet repressors with recognition specificity for the tetO-4C5G operator variant.Gene. 2007 Dec 1;404(1-2):93-100. doi: 10.1016/j.gene.2007.09.002. Epub 2007 Sep 12. Gene. 2007. PMID: 17928170
-
Structural basis of gene regulation by the tetracycline inducible Tet repressor-operator system.Nat Struct Biol. 2000 Mar;7(3):215-9. doi: 10.1038/73324. Nat Struct Biol. 2000. PMID: 10700280
-
Gene regulation by tetracyclines. Constraints of resistance regulation in bacteria shape TetR for application in eukaryotes.Eur J Biochem. 2003 Aug;270(15):3109-21. doi: 10.1046/j.1432-1033.2003.03694.x. Eur J Biochem. 2003. PMID: 12869186 Review.
-
Mechanisms underlying expression of Tn10 encoded tetracycline resistance.Annu Rev Microbiol. 1994;48:345-69. doi: 10.1146/annurev.mi.48.100194.002021. Annu Rev Microbiol. 1994. PMID: 7826010 Review.
Cited by
-
The complex formed between a synthetic RNA aptamer and the transcription repressor TetR is a structural and functional twin of the operator DNA-TetR regulator complex.Nucleic Acids Res. 2020 Apr 6;48(6):3366-3378. doi: 10.1093/nar/gkaa083. Nucleic Acids Res. 2020. PMID: 32052019 Free PMC article.
-
Synthetic mammalian trigger-controlled bipartite transcription factors.Nucleic Acids Res. 2013 Jul;41(13):e134. doi: 10.1093/nar/gkt405. Epub 2013 May 17. Nucleic Acids Res. 2013. PMID: 23685433 Free PMC article.
-
The crystal structure of AcrR from Mycobacterium tuberculosis reveals a one-component transcriptional regulation mechanism.FEBS Open Bio. 2019 Oct;9(10):1713-1725. doi: 10.1002/2211-5463.12710. Epub 2019 Aug 20. FEBS Open Bio. 2019. PMID: 31369208 Free PMC article.
-
NO-Stressed Y. pseudotuberculosis Has Decreased Cell Division Rates in the Mouse Spleen.Infect Immun. 2022 Aug 18;90(8):e0016722. doi: 10.1128/iai.00167-22. Epub 2022 Jul 11. Infect Immun. 2022. PMID: 35862700 Free PMC article.
-
Antimicrobial resistance (AMR) nanomachines-mechanisms for fluoroquinolone and glycopeptide recognition, efflux and/or deactivation.Biophys Rev. 2018 Apr;10(2):347-362. doi: 10.1007/s12551-018-0404-9. Epub 2018 Mar 10. Biophys Rev. 2018. PMID: 29525835 Free PMC article. Review.
References
-
- Ohno S. Evolution by Gene Duplication. New York: Springer; 1970.
-
- Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. - PubMed
-
- Lewis M, Chang G, Horton NC, Kercher MA, Pace HC, Schumacher MA, Brennan RG, Lu P. Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science. 1996;271:1247–1254. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
